 News
News  Market Data
Market Data  Discover
Discover 
Support: 888-992-3836
Copyright © 2023 InvestorsHub Inc.


Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
Far from fair. Nothing post hoc about the design. The author should have questioned that remark or got Liau’s take on it. Because it is not.
This article is actually pretty fair and gives both sides a fair hearing in the traditional fashion of objective journalism. We’d prefer just a “DCVax-L is awesome!” but a real journalist will not be pumping even if they feel that Dr. Liau is probably right as seems to be the case here.
One link includes mention of the upcoming trial that will combine ATL-DC, PLX3397 and pembrolizumab.
The link that mentions the trial was updated a couple months ago, so I would take that as current.
The trial has not yet been posted on ct.gov, so no idea when, or if, it runs. There are some trials running that combine pembrollizumab and PLX3397.
Wolves can eat shark! ;) #NWBO

Another NWBO article showed up on my news feed this morning.
https://apple.news/AGz2Q28YbQtuOnPCfB83rxQ
Excellent posts by you and Senti!
I tried Google translate on that one and i got:

Larppis
Re: ATLnsider post# 539288
Sunday, November 27, 2022 12:41:29 AM
Post#
539289
of 539352
There are actually two different sites to read about the SPORE projects. On the other one they specifically mention Merck and Pembrolizumab (Keytruda), on the other one they do not.
This one has the mention:
https://trp.cancer.gov/spores/abstracts/ucla_brain.htm
This one does not:
https://cancer.ucla.edu/research/ucla-brain-spore/research-projects
ATLnsider
Re: biosectinvestor post# 539285
Sunday, November 27, 2022 12:31:54 AM
Post#
539288
of 539351
Also, I don’t believe the UCLA Spore project actually specified Keytruda would be used in the combo trial. I only see it saying: PD-1 mab
As you may know, “mab” is the suffix ending for all PD-1 inhibitors, and it includes BMY’s product Opdivo (nivolumab).
Here is the UCLA Spore project info:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3071163/
Logo of nihpa
Clin Cancer Res. Author manuscript; available in PMC 2012 Mar 15.
Published in final edited form as:
Clin Cancer Res. 2011 Mar 15; 17(6): 1603–1615.
Published online 2010 Dec 6. doi: 10.1158/1078-0432.CCR-10-2563
PMCID: PMC3071163
NIHMSID: NIHMS257161
PMID: 21135147
Gene expression profile correlates with T cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy
Robert M. Prins,1,4,5,* Horacio Soto,1 Vera Konkankit,1 Sylvia K. Odesa,1 Ascia Eskin,2 William H. Yong,3 Stanley F. Nelson,2,4,5 and Linda M. Liau1,4,5
Author information Copyright and License information Disclaimer
The publisher's final edited version of this article is available free at Clin Cancer Res
Associated Data
Supplementary Materials
Go to:
Abstract
Purpose
To assess the feasibility, safety, and toxicity of autologous tumor lysate-pulsed dendritic cell (DC) vaccination and toll-like receptor (TLR) agonists in patients with newly diagnosed and recurrent glioblastoma. Clinical and immune responses were monitored and correlated with tumor gene expression profiles.
Experimental Design
Twenty-three patients with glioblastoma (WHO grade IV) were enrolled in this dose-escalation study and received three biweekly injections of glioma lysate-pulsed DCs followed by booster vaccinations with either imiquimod or poly-ICLC adjuvant every three months until tumor progression. Gene expression profiling, IHC, FACS, and cytokine bead arrays were performed on patient tumors and PBMC.
Results
DC vaccinations are safe and not associated with any dose-limiting toxicity. The median overall survival from the time of initial surgical diagnosis of glioblastoma was 31.4 months, with a one-, two-, and three-year survival rate of 91%, 55% and 47%, respectively. Patients whose tumors had mesenchymal gene expression signatures exhibited increased survival following DC vaccination compared to historical controls of the same genetic subtype. Tumor samples with a mesenchymal gene expression signature had a higher number of CD3+ and CD8+ tumor infiltrating lymphocytes (TILs) compared with glioblastomas of other gene expression signatures (p = 0.006).
Conclusion
Autologous tumor lysate-pulsed DC vaccination in conjunction with TLR agonists is safe as adjuvant therapy in newly diagnosed and recurrent glioblastoma patients. Our results suggest that the mesenchymal gene expression profile may identify an immunogenic subgroup of glioblastoma that may be more responsive to immune-based therapies.
Go to:
INTRODUCTION
Glioblastoma is a lethal malignant brain tumor with overall survival rates of less than 3.3% at 5 years (1). Glioblastoma remains one of the diseases for which there is no curative therapy. Despite advances in the identification of potential targets for glioma therapy and recent clinical trials utilizing biological therapies and newer cytotoxic agents (2–4), the prognosis of patients with primary malignant brain tumors remains dismal. This sobering fact underscores the need to rethink conventional approaches to the treatment of malignant brain tumors and to base therapeutic strategies on continuing advances in our knowledge of tumor biology and immunology.
The potential therapeutic benefit of eliciting an anti-tumor immune response in cancer patients was first suggested decades ago. Immunotherapy is theoretically appealing because it offers the potential for a high degree of tumor-specificity, while sparing normal brain structures (5). One such approach uses professional antigen-presenting cells, known as dendritic cells (DC), co-cultured with autologous tumor lysate to immunologically target endogenous tumor antigens. Initial studies of DC-based vaccine therapy for malignant gliomas have shown acceptable safety and toxicity profiles (6–14), and multi-center randomized Phase II and III studies are currently underway.
Previous pre-clinical studies (15, 16) strongly suggested that toll-like receptor (TLR) agonists (e.g., imiquimod, poly ICLC), could enhance dendritic cell activation and migration, as well as stimulate T cell-mediated anti-tumor immune responses in murine glioma models. To translate these findings, a Phase I clinical trial was initiated to evaluate the adjunctive use of DC vaccination with TLR agonists for its feasibility, safety, and toxicity in patients with newly diagnosed and recurrent glioblastoma. Herein, we report the results of this Phase I clinical trial, together with immune monitoring data and novel correlative studies associating overall survival with gene expression signatures and increased tumor infiltrating lymphocytes for the glioblastoma patients.
Go to:
PATIENTS AND METHODS
Patient eligibility
This phase I clinical trial was approved by the UCLA IRB and registered with the NCI as NCT00068510. Written informed consent was obtained from all patients. Inclusion criteria were: newly diagnosed or recurrent glioblastoma (WHO Grade IV) that were amenable to surgical resection, a Karnofsky performance score (KPS) = 60%, evidence of normal bone marrow function (e.g., hemoglobin = 9 g/dL, absolute granulocyte count = 1,500/µl and platelet count = 100,000 K), adequate liver function (SGPT, SGOT, and alkaline phosphatase = 2.5 times upper limit of normal; and bilirubin = 1.5 mg/dL), and adequate renal function BUN or creatinine = 1.5 times institutional normals) prior to starting therapy. Exclusion criteria included allergies to any components of the DC vaccine, concurrent or prior corticosteroid use within 10 days of initial vaccination, the presence of acute infection requiring active treatment, unstable or severe intercurrent medical conditions (e.g., pulmonary, cardiac, or other systemic disease), known immunosuppressive disease, positive serology for HIV or hepatitis B, history of an autoimmune disease, or prior history of other malignancies.
Preparation of Autologous Tumor Lysate
Fresh tumor samples from surgical resection were transported under sterile conditions to the UCLA-Jonsson Cancer Center GMP facility and used to generate autologous tumor lysate, as previously described (8, 17). Tumor tissue was minced, digested in collagenase (Advanced Biofactures, Lynbrook, NY) and Dnase-1 (Dornase-a, Genentech, San Francisco, CA) for 8–12 hours at room temperature. To generate lysates, tumor cell suspensions were subjected to five freeze-thaw cycles, centrifuged for 10 minutes at 800×g, and the cell-free supernatants were obtained. Protein concentrations of each tumor lysate were determined using a Bio-Rad DC protein assay (Bio-Rad Corp., Hercules, CA), and lysates with 100 µg of measured protein were used to pulse DC for each injection.
Preparation of Autologous Dendritic Cells and pulsing with glioma lysate
Monocyte-derived DCs were established from adherent peripheral blood mononuclear cells (PBMC) obtained via leukapheresis performed at the UCLA Hemapheresis Unit. Blood was additionally drawn as a source of autologous serum for the DC cultures. All ex vivo DC preparations were performed in the UCLA-Jonsson Cancer Center GMP facility under sterile and monitored conditions. Dendritic cells were prepared by culturing adherent cells from peripheral blood in RPMI-1640 (Gibco) and supplemented with 10% autologous serum, 500 U/mL GM-CSF (Leukine®, Amgen, Thousand Oaks, CA) and 500 U/mL of IL-4 (CellGenix), using techniques described previously (8). Following culture, DCs were collected by vigorous rinsing and washed with sterile 0.9% NaCl solution. The purity and phenotype of each DC lot was also determined by flow cytometry (FACScan flow cytometer; BD Biosciences, San Jose, CA). Cells were stained with FITC-conjugated CD83, PE-conjugated CD86 and PerCP-conjugated HLA-DR mAb’s (BD Biosciences). Release criteria were >70% viable by trypan blue exclusion, and >30% of the large cell gate being CD86+ and HLA-DR+. One day before each vaccination, DC were pulsed (co-cultured) with 100 µg of tumor lysate overnight, washed, and the final product was tested for sterility by Gram stain, mycoplasma and endotoxin testing prior to injection.
Treatment Schema
Newly diagnosed glioblastoma patients underwent surgery and a standard course of external beam radiotherapy with concurrent temozolomide chemotherapy prior to DC vaccination (4). These patients were given 3 biweekly DC vaccinations following standard chemo-radiation and prior to adjuvant temozolomide treatment. Recurrent glioblastoma patients had previous radiation therapy and chemotherapy prior to presenting with tumor recurrence, so they underwent surgical resection of their tumors followed by DC immunotherapy after they had recovered from surgery and were tapered off peri-operative steroids. This ranged from 7–30 weeks after surgery.
Vaccine Administration
On the day of each DC vaccination, a 1 ml vaccine dose was drawn into a sterile tuberculin syringe and administered as an intradermal (i.d.) injection (using a 25-gauge needle) in the arm region below the axilla, with the side of administration rotated for each vaccination. Subjects were monitored for two hours post-immunization in the UCLA General Clinical Research Center (GCRC). Eligible patients initially received three (3) intradermal injections at biweekly intervals. If patients did not develop any toxic side effects from the experimental treatment and had stable disease for over three months, they received booster injections at the same dosage of tumor lysate-pulsed DC concurrently with either 5% imiquimod cream (Aldara™, a TLR-7 agonist) or poly-ICLC (Hiltonol™, a TLR-3 agonist). Due to initial safety/toxicity concerns of experimental allergic encephalomyelitis (EAE) (18) resulting from the combined use of DC vaccination and TLR agonists, these immune response modifiers were used only in the booster phase of the protocol, after patients had shown acceptable toxicity profiles to DC-lysate vaccinations alone. Booster vaccinations were given at 3 month intervals in between 28-day cycles (5 days on/23 days off) of temozolomide for up to 10 boosters or until tumor progression. For those receiving imiquimod as adjuvant, patients applied 5% imiquimod cream topically over the DC vaccination site one day prior to each vaccination cycle, immediately after DC vaccination, and then daily for an additional three days post-vaccination. For patients in the poly-ICLC cohort, intramuscular (i.m.) injections of 20 µg/kg of poly-ICLC were administered immediately prior to each DC injection at the vaccine injection site. All patients had a baseline brain MRI scan within one month prior to starting the immunotherapy and every 2 months thereafter or when clinically indicated.
Patient Assessment
Toxicity was monitored and graded according to the National Cancer Institute (NCI) Common Toxicity Criteria. The overall incidence of adverse events was recorded. Neurological exams were performed before and 30 minutes after each vaccination, as well as at all follow-up visits. Time to tumor progression (TTP) was defined as the interval from surgical resection until the first observation of tumor progression, as evidenced by magnetic resonance imaging (MRI) or clinical deterioration. Tumor progression was also considered to be non-reversible neurologic progression, permanently increased steroid requirement (applies to stable disease only), or early discontinuation of treatment. Overall survival (OS) time was determined from the date of surgery at the time of initial diagnosis of glioblastoma to date of death.
Flow Cytometry and Cytometric Bead Array
PBMC from patients enrolled on this clinical trial (pre- and post-vaccination) and PBMC from normal volunteers were thawed in warmed RPMI+2% FBS, washed and stained for the expression of CD3, CD4 and CD25 (all from BD Biosciences; San Diego, CA), followed by the intracellular labeling of Foxp3 (eBioscience; San Diego, CA). Stained cells were acquired on a BD FacsCalibur flow cytometer and analyzed using FloJo software. The frequencies of CD3+CD4+Foxp3+ and CD3+CD4+CD25+Foxp3+ PBMC’s were compared. For cytokine analysis, serum from patients enrolled on this clinical trial was thawed and incubated with the Cytometric Bead Array (CBA) Human Th1/Th2 Capture Beads (BD Biosciences), washed and subjected to analysis on a BD FacsCalibur flow cytometer together with cytokine standards. Quantitative assessment of cytokine levels was accomplished with a Microsoft Excel-based CBA software program.
Immunohistochemical (IHC) staining
Serial paraffin sections of pre-treatment tumor specimens were cut to 3 µm thickness and stained with anti-human antibodies against CD3 (DakoCytomation; Carpinteria, CA) and CD8 (DAKO Corp.; Carpinteria, CA). Sections were baked for 1 hour at 60°C, deparaffinized, and endogenous peroxidase activity quenched by treating with 0.5% H2O2 in methyl alcohol for 10 minutes. Heat-induced epitope retrieval was performed on the slides using 0.01 M citrate buffer, pH=6.0 (for CD3, CD8) in a vegetable steamer (Black & Decker); slides were heated for 25 minutes, cooled, and washed in 0.01 M phosphate buffered saline. All slides then were placed on a DAKO Autostainer (DAKO Corp.) and then sequentially incubated in primary antibody for 30–60 minutes, then rabbit anti-mouse secondary immunoglobulins (DAKO Corp.) for 30 minutes. Diaminobenzidine and hydrogen peroxide were used as the substrates for the peroxidase enzyme. For the negative controls, mouse isotype or rabbit immunoglobulins (DAKO Corp.) were used in place of the primary antibodies. Positive labeling was evaluated and scored by a board-certified neuro-pathologist (WHY) in a blinded fashion.
Microarray Studies
Of the twenty-three glioblastoma patients, sixteen patients had sufficient residual tumor tissue for microarray molecular analysis at the end of the trial. Total RNA was purified from pre-treatment, fresh frozen tumor samples using the RNeasy mini kit (Qiagen) and collected as part of the IRB-approved research protocol. cRNA was generated, quantified and hybridized to U133 Plus 2.0 arrays at the UCLA DNA Microarray Facility using standard Affymetrix protocols. CEL files were normalized using the Celsius Microarray Database (19), with robust multichip average (RMA) from Bioconductor (version 2.10) relative to 50 samples of the same platform. The Hierarchical Clustering (HC) classification for each glioma was determined by a gene voting strategy as described previously (20, 21). Briefly, the mean value of each probeset was evaluated from all samples within the U133 Plus 2.0 platform using the 377 gene probeset list and assigned to a HC group (21). Tumors were assigned to a HC group when the number of probes above the normalized mean was greater than 30% of a given probeset. The overall survival of patients tumors on this Phase I clinical trial was compared with the overall survival of patients from a collection of samples previously assigned to HC groups (21).
Statistical Analysis
Time to tumor progression (TTP) and overall survival (OS) curves were determined using the Kaplan-Meier method. The Log-rank (Mantel-Cox) test was used to compare curves between study and control groups. All P-values are two-tailed, and p < 0.05 was considered statistically significant. Statistics were analyzed using GraphPad Prism software.
Go to:
RESULTS
Patient characteristics
Twenty-three patients with histologically proven WHO grade IV (glioblastoma) were enrolled in this protocol (Table 1). Fifteen had newly diagnosed tumors, while eight had recurrent disease. There were sixteen men and seven women, with an age range of 26 to 74 years (mean age of 51 years).
Table 1
Patient Characteristics
Patient
ID Tumor
Pathology Age Gender KPS OS
(mo.) HC Type Dose
(106) Adjuvant Pre-vacc. Tx. Post-vacc. Tx. Related Adverse Events
GBM1-1 GBM 39 M 90 33.83 * 1 Imiquimod temozolomide temozolomide, isoretinoin, celecoxib, Reoperation, SRS* Fatigue, nausea/vomiting, diarrhea
GBM1-2 GBM 39 M 90 >88.87 Mes 1 Imiquimod temozolomide temozolomide, isoretinoin, CCNU, Gliadel™ Fatigue, arthralgia, low-grade fever
GBM1-3 GBM 34 M 90 >91.3 Mes 1 Imiquimod temozolomide temozolomide, isoretinoin Lymphadenopathy, injection site reaction, low-grade fever, myalgia
GBM1-4 rec. GBM 61 M 70 18.57 * 1 None temozolomide, thalidomide, isoretinoin, Newcastle virus
GBM1-5 GBM 58 M 70 10.3 Pro 1 None temozolomide irinotecan, bevacizumab
GBM1-6 GBM 63 F 80 >37.6 Mes 1 Imiquimod temozolomide temozolomide Shingles
GBM1-7 rec. GBM 41 F 80 10.93 Mes 1 None irinotecan, bevacizumab irinotecan, bevacizumab
GBM1-8 rec. GBM 34 M 100 >40.5 PN 1 Poly ICLC erlotinib, temozolomide, ANG, CCNU, celecoxib, tamoxifen, CCNU, celecoxib, tamoxifen,
GBM1-9 GBM 50 M 90 >9.03 * 1 Poly ICLC temozolomide
GBM5-1 GBM 40 F 80 17.97 * 5 None temozolomide, isoretinoin CCNU, gefitinib, rapamycin, carboplatin Injection site reaction
GBM5-2 rec. GBM 54 M 80 17.3 * 5 None temozolomide, isoretinoin, CCNU Carboplatin Allergic rhinitis, itching at injection site, pruritis
GBM5-3 GBM 26 M 90 81.4 PN 5 Imiquimod temozolomide, isoretinoin, CCNU carboplatin, irinotecan, bevacizumab, dasatanib, simvastatin, rosiglitazone, procarbazine, CCNU, cyclophosphamide Injection site itching, dryness and pruritus, lymphadenopathy, nausea, diarrhea, vomiting, fatigue
GBM5-4 GBM 43 M 90 >59.0 Mes 5 Imiquimod temozolomide temozolomide
GBM5-5 GBM 45 F 90 34.97 PN 5 None temozolomide irinotecan, bevacizumab, CCNU, erlotinib
GBM5-6 rec. GBM 53 M 80 22.33 Mes 5 Poly ICLC temozolomide, Gliadel™ irinotecan, bevacizumab Nausea, heartburn, constipation
GBM10-1 rec. GBM 58 F 70 28.93 PN 10 None temozolomide, irinotecan, paclitaxel Headaches, nausea/vomiting
GBM10-2 GBM 70 F 100 23.0 * 10 None temozolomide p-EGFR/p-ErbB2 inhib, everolimus, carboplatin, bevacizumab, CCNU
GBM10-3 GBM 50 M 90 36.33 * 10 Imiquimod temozolomide irinotecan, bevacizumab Dermatitis, rash,
GBM10-4 GBM 59 M 80 52.6 Pro 10 Imiquimod temozolomide, isoretinoin irinotecan, bevacizumab, carboplatin, CCNU Anorexia, abdominal pain,
GBM10-5 GBM 64 M 90 13.63 Mes 10 None temozolomide bevacizumab, CCNU
GBM10-6 GBM 66 M 90 37.73 Mes 10 Imiquimod temozolomide irinotecan, bevacizumab, CCNU, etoposide, procarbazine, tamoxifen Fatigue, left shoulder pain/arthralgia
GBM10-7 rec. GBM 74 F 60 17.07 Mes 10 None temozolomide temozolomide
GBM10-8 rec. GBM 52 M 60 16.23 PN 10 None temozolomide Upper lip blisters, rash
Open in a separate window
*SRS = stereotactic radiosurgery
DC preparation and phenotype
DCs were generated from adherent PBMC cultured in the presence of 500 U/mL GMP-grade IL-4 and 500 IU/mL of GM-CSF for one week prior to harvest, as reported previously (8). All final autologous tumor lysate-pulsed DC preparations consistently contained a high percentage of viable large, granular cells and were free of contamination. Our DC preparations expressed high levels of MHC class I (HLA-A,B,C), MHC class II (HLA-DR), B7.2 costimulatory molecule (CD86), and CD40, but lower expression of CD14 and CD80 (Supplementary Table 1). These DC preparations were partially mature, with <45% of the large cells expressing HLA-DR and CD83, as might be expected for a protocol without a dedicated maturation step. Overnight incubation with tumor lysates induced some DC maturation, as evidenced by an increase in the median fluorescence intensity (MFI) of CD83 (data not shown), similar to previously reported findings (22).
Safety and toxicity
DC vaccinations were well-tolerated, with no major adverse events (NCI Common Toxicity Criteria grade 3 or 4) observed in any subject during the vaccine cycles (Table 1). There were no clinical or radiological signs of EAE or other autoimmune reactions in any patient. There were anecdotal cases of transient increased T2/FLAIR and enhancing lesions on MRI after DC vaccination, which may have suggested inflammatory responses post DC vaccination, particularly in the mesenchymal gene-clustered cohort of patients (Fig. 1). However, these MRI changes resolved in due course and did not require surgical intervention. The appearance and disappearance of such MRI findings, presumed to be related to vaccination and neuroinflammation, was noted in three of our patients (GBM 1–2, 1–3, and 5–4). These three patients were in the mesenchymal subgroup and are still alive over five years from the initial diagnosis of glioblastoma. Nausea/vomiting, headache and fatigue, diarrhea, low-grade fever and and pain/itching at the injection site were the most common symptoms associated with the treatment (Table 1). Local lymphadenopathy was observed in one patient, temporally coinciding with the expansion of HCMV-specific T cell expansion (23). In patients who concomitantly received 5% imiquimod cream or poly-ICLC with DC vaccination in the booster phase, no new additional toxicities were reported. Two patients consistently reported transient fevers (=103° F) with each DC + poly-ICLC injection. Cumulatively, these data suggest a low toxicity profile for autologous tumor lysate-pulsed DC plus TLR agonists at all DC dose levels tested.
An external file that holds a picture, illustration, etc.
Object name is nihms257161f1.jpg
Figure 1
MRI changes after DC vaccination. Transient increase in MRI T2/FLAIR lesions (A) and contrast enhancement (B) observed in a primary, newly diagnosed glioblastoma patient following DC vaccination (patient GBM5-4). Axial T2/FLAIR (A) and T1/contrast (B) MRI scans taken at 2 weeks pre-vaccination, 2 weeks post-vaccination, and 4 months later.
Systemic cytokine responses and regulatory T-cell populations following DC vaccination with TLR agonists
Others have assessed systemic immune responsiveness from autologous tumor lysate-pulsed DC vaccination by either delayed type hypersensitivity skin testing (DTH) (6, 12) or by restimulating PBMC with lysate-pulsed DC in vitro, followed by assessment of interferon-gamma (IFN-?) (10, 12). However, the correlations with clinical outcome have not been consistent. In this trial, we elected to assess for more global systemic cytokine responses and changes in regulatory T cell (Treg) frequency that may be induced by our vaccination strategy.
Peripheral blood changes in the frequency of CD3+CD4+Foxp3+ T cells were compared prior to and after DC vaccination for patients with available pre and post-treatment PBMC. We observed that glioblastoma patients on this clinical trial possessed increased frequencies of peripheral blood CD3+CD4+Foxp3+ or CD3+CD4+CD25+Foxp3+ lymphocytes compared with normal volunteers (Fig. 2A). However, at the time points measured, there were no relevant changes in the frequency of this lymphocyte population after immunotherapy that statistically correlated with clinical outcome (data not shown).
An external file that holds a picture, illustration, etc.
Object name is nihms257161f2.jpg
Figure 2
Peripheral blood immune monitoring data. (A) PBMC’s from normal volunteers and DC trial patient pre-vaccination timepoints were thawed and stained for the expression of CD3, CD4 and CD25, followed by the intracellular labeling of Foxp3. Stained cells were acquired on a BD FacsCalibur flow cytometer and analyzed using FloJo software. The frequencies of CD3+CD4+Foxp3+ and CD3+CD4+CD25+Foxp3+ PBMC’s between normal volunteers and glioblastoma patients enrolled in this trial are compared. (*p=0.04; **p=0.01) (B,C) Serum cytokine responses, measured pre- and day 14 post-vaccination, after the initial course of DC vaccination (B) or after booster DC vaccinations with either 5% imiquimod or poly ICLC (C). Serum from patients enrolled on this clinical trial was thawed, labeled with cytometric bead array (CBA) antibody-coated beads, washed and subjected to analysis on a BD FacsCalibur flow cytometer together with cytokine standards. Quantitative assessment of cytokine levels was accomplished with a Microsoft Excel-based CBA software program. (D) Th1/Th2 cytokine ratios. Raw cytokine data for serum TNF-a and IL-10 at each timepoint were divided to generate a Th1:Th2 ratio.
To assess the cytokine microenvironment after DC vaccination with and without the addition of TLR agonists, we performed cytometric bead arrays from patient serum during the time course of the clinical trial to evaluate Th1 and Th2-type cytokine levels. Detectable increases in serum TNF-a and IL-6 were observed after DC vaccination (Fig. 2B, Suppl. Fig. 1A). However, the serum cytokine levels were variable between patients and the magnitude of changes did not seem to correlate with clinical outcome. Log-fold elevations in serum TNF-a and IL-6 were observed after booster DC vaccinations with either 5% imiquimod cream or 20 µg/kg poly ICLC (Fig. 2C, Suppl. Fig. 1B). To assess whether the Th1/Th2 cytokine balance might be relevant, we calculated ratios of each Th1-type cytokine with Th2-type cytokines to generate an effector/regulatory cytokine ratio (Fig. 2D). However, such information was also not significantly associated with the clinical outcome (data not shown), although our sample numbers may have been too small to detect statistical significance.
Dose escalation
A typical dose escalation scheme was performed with autologous tumor lysate-pulsed DC vaccination, using 1, 5 and 10 million DC administered intradermally. A fixed amount of lysate (100 µg) was added to the DC and incubated overnight prior to injection. The patient characteristics and survival data for each dose cohort are outlined in (Supplementary Table 2). In this dose escalation trial, there was no relationship between increasing DC dose and toxicity or specific adverse events of any kind. There were also no DC dose-dependent differences in immunological responses tested. As seen in Supplementary Table 2, the median overall survival was actually longer in the 1 million DC dose cohort compared with the higher dose cohorts. However, these differences in OS were not statistically significant, given the small sample size in each dose cohort and age differences between groups.
Survival analysis
Although this Phase I clinical trial was not powered to detect clinical efficacy, tumor response was monitored by clinical and MRI assessments at baseline (within one month prior to therapy), and every eight weeks thereafter as surrogate markers for clinical response and tumor status. Objective clinical data are summarized below and are listed in Table 1. When considering all 23 glioblastoma patients enrolled in this clinical trial (newly diagnosed and recurrent patients), the median time to tumor progression (TTP) was 15.9 months. The median overall survival time (OS), taken from the date of initial surgical diagnosis of glioblastoma, was 31.4 months. Overall survival from the time of initial diagnosis at one, two and three years was 91%, 55% and 47%, respectively. If we include only those who received the DC vaccine in the newly diagnosed setting (n=15), the median overall survival is 35.9 months, with a mean follow-up time of over four years, and one, two and three-year survival rates of 93%, 77% and 58%, respectively. For recurrent patients that enrolled in our vaccine trial (n=8), the median overall survival was 17.9 months from the time of initial glioblastoma diagnosis. OS was significantly longer for those who received DC vaccination at initial diagnosis compared to those who enrolled in this trial at the time of recurrence (p=0.03; Supplementary. Fig. 2).
Microarray gene expression profiling
Since gene expression patterns have been shown to be highly correlated with survival in various cancers, we investigated whether the genetic signature of glioblastomas (20) was associated with clinical outcome in this DC immunotherapy trial. In patients where available pre-treatment tumor samples were available, we performed microarray-based gene expression classification as previously published (20, 21). As shown in Figure 3, gene expression profiling of our pre-treatment tumor samples produced the typical proneural (PN), proliferative (Pro) and mesenchymal (Mes) hierarchical clusters, using probesets previously described by our group (20, 21). Furthermore, we validated these hierarchical clusters using the UCSF-Genentech and TCGA probesets (24, 25), which yielded similar gene expression signatures for our DC lysate patients (data not shown).
An external file that holds a picture, illustration, etc.
Object name is nihms257161f3.jpg
Figure 3
Microarray-based, expression profiling of pre-treatment glioblastoma samples from DC vaccine patients. Total RNA was isolated from frozen, surgically-resected tumors and subjected to global gene expression classification using Affymetrix human U133 Plus 2.0 microarray chips. Sufficient fresh-frozen tissue was available for extraction of high-quality RNA (without amplification) in 17 of the cases. Proneural (HC1, yellow legend), Proliferative (HC2A, blue legend), and Mesenchymal (HC2B, red legend) gene expression signatures were identified using probesets previously published (21). Heat maps were created using the dChip microarray software program.
The mesenchymal gene expression signature is defined by overexpression of many inflammatory-associated genes. Thus, we hypothesized that there might be a difference in the clinical outcome of patients on our trial that could be linked to the local microenvironment of the original tumor. In order to control for any selection bias that might have been introduced by the requisite eligibility criteria for patients receiving the DC vaccine (i.e., subjects needing to be alive and off steroids long enough for vaccine preparation and administration), we eliminated any control patients that died within ~250 days of initial diagnosis for the purposes of our comparative analysis. We also stratified for patients who received radiation alone vs. radiation plus concurrent temozolomide chemotherapy after initial surgical resection, and found no statistical difference in these two groups when the early progressors (OS<250 days) were eliminated. As shown in Figure 4, patients enrolled on our trial with the proneural gene expression signature had an overall survival that was indistinguishable from a set of 60 contemporary proneural tumors analyzed from UCLA and three other institutions (21) (p=0.664; Fig. 4A). In contrast to this, patients in our DC vaccine trial with mesenchymal gene expression signatures had a significantly extended survival compared with 82 concurrently collected tumors that were found to have these same gene expression signatures (p=0.0046; Fig. 4B). While these data are not intended to represent efficacy, such information is noteworthy because glioblastoma patients with mesenchymal gene expression patterns typically have the worst prognosis and are the most refractory to current therapies (21, 24, 25).
An external file that holds a picture, illustration, etc.
Object name is nihms257161f4.jpg
Figure 4
Extended survival in DC vaccinated patients with mesenchymal gene expression signatures, but not in patients with a proneural signature. The overall survival time of DC vaccine patients expressing a (A) Proneural (PN) gene signature (n=5) or (B) Mesenchymal (Mes) gene signature (n=9) was compared with the survival generated from a control, multi-institutional dataset of PN (n=60) or Mes glioblastomas (n=82; solid lines) previously published by our group (21). To accurately account for the potential bias associated with the time delay needed to generate the DC vaccine, we omitted control patients that experienced early progression (<250 days). PN comparison: p=0.664 (not statistically different, ns); Mes comparison (p=0.0046) by the Log-rank (Mantel-Cox) Test calculated in GraphPad software.
Gene expression signature and tumor-infiltating lymphocytes
The density and location of T lymphocyte accumulation within certain solid tumors have been associated with extended survival (26, 27), and recent evidence suggests that such a correlation may exist in malignant glioma (28). However, an association with the subtype of tumor or treatment modality has not been addressed.
Since the mesenchymal expression signature includes numerous genes associated with inflammation, and tumor-specific T cells are known to be attracted to pro-inflammatory signals, we evaluated whether patients on our DC trial with mesenchymal gene expression signatures also had increased tumor infiltrating lymphocytes (TILs). As shown in Figure 5, tumors with a mesenchymal gene expression signature had significantly increased CD3+ and CD8+ TILs compared with PN tumors (p=0.006). Although our sample size is small, we also found qualitatively increased CD3+ and CD8+ TIL density after DC vaccination in tumors resected at recurrence (Fig. 5B). In post-DC vaccinated tumors resected/biopsied at the time of recurrence, increases in CD3+ and CD8+ TILs were associated with the mesenchymal gene expression profile, but not necessarily with the dose of DC given. Such findings point to a potential mechanism by which distinct glioblastoma tumor subtypes might be differentially responsive to immune-based therapies.
An external file that holds a picture, illustration, etc.
Object name is nihms257161f5.jpg
Figure 5
Increased density of CD3+ and CD8+ lymphocytes in Mes gene expression groups compared with PN tumor sections. (A) 3 µm paraffin-embedded adjacent tissue sections from DC vaccinated patients were stained separately with CD3 and CD8 antibodies and scored in a blinded fashion by a neuropathologist (WHY). The IHC scores were compared between samples known to be PN (n=5) vs. Mes tumor samples (n=9). *p=0.006 by two-tailed t test calculated in GraphPad software. (B) Representative hematoxylin & eosin staining and CD8 IHC staining (pre- and post-DC vaccination) of a PN and Mes glioblastoma showing increased CD8+ TILs in the Mes glioblastoma. Original magnification: × 400.
Go to:
DISCUSSION
In this Phase I study, we report the safety, feasibility, and bioactivity of a vaccine comprised of autologous DC pulsed with autologous tumor lysate as an adjuvant following surgical resection with standard chemo-radiotherapy. Unlike our previous reported DC vaccination strategy (8) and those reported by other groups (6, 9–13, 29), we included “booster” vaccinations with the innate immune response modifiers, 5% imiquimod (Aldara™) or poly-ICLC (Hiltonol™) based on our pre-clinical studies suggesting that pro-inflammatory innate immune signals could enhance DC activation, trafficking to lymph nodes, and the priming of anti-tumor antigen-specific T lymphocytes (15). There were no dose-limiting toxicities and no detectable differences in safety or efficacy among the three DC dose levels tested. Of note, there was a significant difference in the average age of patients in the 10 million DC cohort compared to the other dose cohorts, which could influence the difference in overall survival. However, another possible hypothesis is that this trend in the data was a reflection of a dilutional decrease in antigens available for presentation by DC at the highest DC dose cohort (10×106 cells), given that the quantity of lysate was fixed (at 100 µg per dose) despite the increased DC cell dose.
The concomitant administration of 5% imiquimod or poly ICLC with DC vaccination was also found to be safe and did not result in any additional toxicity or adverse events. To our knowledge, this is the first report of the use of TLR agonists in conjunction with DC vaccination strategies in brain tumor patients. Because TLR agonists were used only in patients in the booster phase, it is unclear whether or to what extent the addition of the TLR agonists contributed to the potential efficacy and overall survival of these patients. Furthermore, imiquimod and poly-ICLC are two different biological agents, targeting different TLRs. Imiquimod activates TLR-7, while poly-ICLC activates TLR-3, but both induce pro-inflammatory cytokine secretion. These complexities make it somewhat difficult to determine how these innate immune modifiers actually contributed to our study endpoints. Nevertheless, this current study establishes the safety of these TLR agonists in conjunction with glioma lysate-loaded DC, and further Phase II studies directly comparing these TLR agonists at the time of initial vaccination (not only in the booster phase) are currently underway.
While the number of glioblastoma patients entered in this Phase I clinical trial was not powered to measure efficacy, the clinical results of this trial are still noteworthy. The median OS from the time of initial surgical diagnosis was 31.4 months for all glioblastoma patients (n=23) treated in this study, including both those enrolled as newly diagnosed and recurrent tumor patients. For those treated in the newly diagnosed setting, the OS was 35.9 months; and the OS was 17.9 months for those who received vaccination at recurrence. In addition, we have had three patients survive over six years to date. Such statistics are compelling in the face of the expected median survival for this disease, which is currently still reported as about 14 months for newly diagnosed patients that receive standard surgery, radiation and temozolomide chemotherapy (4, 30, 31). This compares favorably even when compared with published data for the best clinically defined prognostic group of glioblastoma patients (Recursive Partitioning Analysis, RPA class III: age < 50 years and KPS = 90), whose 2-year survivals were 40% and 29% for RPA III and IV patients, respectively, following treatment with standard radiation and temozolomide (31). Such data is also favorable compared to other recent brain tumor DC-based vaccine trials without booster injections and TLR adjuvants, where the OS was reported as 21.4 months (mean, 11 newly diagnosed and 23 recurrent glioblastoma patients) (10) and 9.6 months (median) in a recurrent glioblastoma population (6).
Glioblastomas are primarily identified by histologic features assigned to cytologically malignant, mitotically active, necrosis-prone tumors established by the World Health Organization (WHO) (32). Such histologic features are generally associated with patient survival, together with performance status, extent of surgical resection, and age. Yet, histologically identical tumors can behave in different ways; a situation that may underlie the biology of this heterogeneous disease. More recently, extensive genetic profiling of these tumors has been able to identify molecularly classifiable subgroups of glioblastoma (i.e., proneural, proliferative, and mesenchymal subtypes),(20, 21, 24, 33–38) which can better predict survival than conventional histopathologic analysis. Such new classification techniques are of interest so that patients can be more appropriately stratified for new treatment strategies (20).
The mesenchymal subgroup of glioblastomas typically have a poorer prognosis than the more common proneural subgroup (21, 24). However, in our study, patients with the mesenchymal gene expression signatures had significantly extended survival compared with a large, multi-institutional cohort (n=82) of glioblastoma samples of the same molecular subgroup treated with various other therapies. No such survival difference was observed in patients from this clinical trial with proneural signatures, compared to other control glioblastoma subjects of the proneural subgroup (n=60). Admittedly, such comparisons with concurrent and historical controls are not meant to imply efficacy, since this Phase I trial did not have a prospectively matched, placebo-controlled arm. Although some prognostic factors, such as age and Karnofsky performance status, were relatively matched in our comparison groups, the extent of surgical resection was not directly compared between the patients in this trial and our concurrent/historical controls. Since we need adequate amounts of tumor (>2 grams) to generate the autologous vaccines, tumor resectability was taken into account in the eligibility criteria. Therefore, it is possible that the extent of surgical resection may have been greater in our DC vaccinated patients compared to concurrent/historical controls, which could have influenced our survival results. Nevertheless, the median OS (31.4 mo.) of our DC-vaccinated patients is still noteworthy, when compared to large series of glioblastoma patients who underwent gross total tumor resections and were treated with concomitant chemo-radiotherapy, where the median survival was reported to be 18.6 months (31).
It is unclear whether the extended survival of our patients with mesenchymal gene expression signatures is a direct result of the vaccine effects, or good responses to follow-up therapies after failing the vaccine. Since mesenchymal signatures represent glioblastoma subgroups that are more resistant to conventional therapy, it can be speculated that DC vaccination somehow makes these tumors more susceptible to subsequent treatments (39). Because adjuvant temozolomide treatment was coordinated into the schedule of the DC booster vaccinations, this is a difficult distinction to make from our study design. Nevertheless, our results do suggest that mesenchymal gene expression signatures express elevated inflammatory gene transcripts and possess an increased density of tumor-infiltrating CD3+ and CD8+ lymphocytes compared with glioblastomas expressing other genetic signatures. As such, we hypothesize that the expression of inflammatory genes (e.g., IL-1R, TNF-a signaling factors, and chemokines) may facilitate the priming and trafficking of tumor-specific T cells into the tumor parenchyma, which might be enhanced by DC vaccination and innate immune response modifiers. Hence, the mesenchymal gene expression signature may have a direct impact on the bioactivity of the vaccine itself, irrespective of post-vaccine therapy. Prospectively designed, randomized, multi-center Phase III clinical trials will be required to validate such hypotheses and proof of clinical benefit remains to be established.
Overall, the results reported here may provide novel insights for prospective patient selection in future immunotherapy studies and lend additional credence for the ability of genetic expression signatures to impart relevant data for personalized cancer treatment. Based on the results of this Phase I trial, we will continue developing more advanced clinical trials with this particular approach. We currently are planning a randomized, multi-center Phase II/III clinical trial of DC vaccination for newly diagnosed glioblastoma (DCVax-Brain™), which will hopefully help to further define which subgroups of patients may respond to tumor vaccination strategies. This in turn may lead to further optimization and refinements of related trials of DC-based vaccines for patients with glioblastoma, with the ultimate goal of developing novel immunotherapeutic strategies for brain cancer patients.
Statement of Translational Relevance
The selective identification of patients who will respond to a particular therapy is of paramount importance, especially for patients diagnosed with malignant glioma. Patients diagnosed with glioblastoma (WHO grade IV) have an expected 5 year survival of less 3.3%. In this study, we report the results of a phase I clinical trial in which glioblastoma patients were treated with a personalized immunotherapy approach, comprised of autologous tumor lysate-pulsed dendritic cell vaccination. In addition, we utilized gene expression profiling to identify a group of patients with a particular gene expression signature (mesenchymal glioblastoma) that had longer survival following DC vaccination, compared to contemporary/historical control patients of the same gene expression subgroup, who did not receive vaccination. This signature was associated with inflammatory transcripts and enhanced tumor infiltrating T lymphocytes. Thus, these results suggest that gene expression signatures may be able to identify an immunogenic subgroup of glioblastoma that could be more responsive to immune-based therapies.
Go to:
Supplementary Material
1
Click here to view.(136K, pptx)
Go to:
Acknowledgments
We thank Timothy Cloughesy, M.D., and Albert Lai, M.D., Ph.D. for helpful discussions and comments.
Research Support. This work was supported in part by NIH/NCI grants K01-CA111402 and RO1-CA123396 (to RMP), R01 CA 112358 (to LML), the Philip R. and Kenneth A. Jonsson Foundations, the Neidorf Family Foundation, the Ben & Catherine Ivy Foundation and Northwest Biotherapeutics, Inc. RMP is the recipient of the Howard Temin NCI Career Development award and STOP Cancer Career Development award. Flow cytometry was performed at the UCLA Jonsson Comprehensive Cancer Center (JCCC) Core Facility and gene expression was performed in the JCCC Gene Expression Shared Resource, which are supported by the NIH award CA16042. Microarray analysis was supported by the Carson Foundation. Tissue acquisition and IHC was supported by the UCLA Brain Tumor Translational Resource (BTTR), while the General Clinical Research Center (GCRC) was supported by M01-RR00865.
Go to:
Footnotes
Disclaimers. The authors do not have any conflict of interests in this work.
Go to:
REFERENCES
1. Deorah S, Lynch CF, Sibenaller ZA, Ryken TC. Trends in brain cancer incidence and survival in the United States: Surveillance, Epidemiology, and End Results Program, 1973 to 2001. Neurosurg Focus. 2006;20:E1. [PubMed] [Google Scholar]
2. Cohen MH, Li Shen Y, Keegan P, Pazdur R. FDA Drug Approval Summary: Bevacizumab (Avastin(R)) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist. 2009 [PubMed] [Google Scholar]
3. Lai A, Filka E, McGibbon B, et al. Phase II Pilot Study of Bevacizumab in Combination With Temozolomide and Regional Radiation Therapy for Up-Front Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Interim Analysis of Safety and Tolerability. Int J Radiat Oncol Biol Phys. 2008 [PubMed] [Google Scholar]
4. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. [PubMed] [Google Scholar]
5. Yang MY, Zetler PM, Prins RM, Khan-Farooqi H, Liau LM. Immunotherapy for patients with malignant glioma: from theoretical principles to clinical applications. Expert Rev Neurother. 2006;6:1481–1494. [PubMed] [Google Scholar]
6. De Vleeschouwer S, Fieuws S, Rutkowski S, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14:3098–3104. [PubMed] [Google Scholar]
7. Kikuchi T, Akasaki Y, Abe T, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother. 2004;27:452–459. [PubMed] [Google Scholar]
8. Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–5525. [PubMed] [Google Scholar]
9. Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008;15:114–121. [PubMed] [Google Scholar]
10. Wheeler CJ, Black KL, Liu G, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68:5955–5964. [PubMed] [Google Scholar]
11. Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. BrJCancer. 2003;89:1172–1179. [PMC free article] [PubMed] [Google Scholar]
12. Yamanaka R, Homma J, Yajima N, et al. Clinical Evaluation of Dendritic Cell Vaccination for Patients with Recurrent Glioma: Results of a Clinical Phase I/II Trial. Clin Cancer Res. 2005;11:4160–4167. [PubMed] [Google Scholar]
13. Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–4979. [PubMed] [Google Scholar]
14. Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–847. [PubMed] [Google Scholar]
15. Prins RM, Craft N, Bruhn KW, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–164. [PubMed] [Google Scholar]
16. Zhu X, Nishimura F, Sasaki K, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10. [PMC free article] [PubMed] [Google Scholar]
17. Yang I, Kremen TJ, Giovannone AJ, et al. Modulation of major histocompatibility complex Class I molecules and major histocompatibility complex-bound immunogenic peptides induced by interferon-alpha and interferon-gamma treatment of human glioblastoma multiforme. J Neurosurg. 2004;100:310–319. [PubMed] [Google Scholar]
18. Bigner DD, Pitts OM, Wikstrand CJ. Induction of lethal experimental allergic encephalomyelitis in nonhuman primates and guinea pigs with human glioblastoma multiforme tissue. J Neurosurg. 1981;55:32–42. [PubMed] [Google Scholar]
19. Day A, Carlson MR, Dong J, O'Connor BD, Nelson SF. Celsius: a community resource for Affymetrix microarray data. Genome Biol. 2007;8:R112. [PMC free article] [PubMed] [Google Scholar]
20. Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–6510. [PubMed] [Google Scholar]
21. Lee Y, Scheck AC, Cloughesy TF, et al. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med Genomics. 2008;1:52. [PMC free article] [PubMed] [Google Scholar]
22. Thumann P, Moc I, Humrich J, et al. Antigen loading of dendritic cells with whole tumor cell preparations. Journal of immunological methods. 2003;277:1–16. [PubMed] [Google Scholar]
23. Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med. 2008;359:539–541. [PMC free article] [PubMed] [Google Scholar]
24. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. [PubMed] [Google Scholar]
25. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. [PMC free article] [PubMed] [Google Scholar]
26. Camus M, Tosolini M, Mlecnik B, et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009;69:2685–2693. [PubMed] [Google Scholar]
27. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. [PubMed] [Google Scholar]
28. Yang I, Tihan T, Han SJ, et al. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci. 2010 [PMC free article] [PubMed] [Google Scholar]
29. Heimberger AB, Sun W, Hussain SF, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol. 2008;10:98–103. [PMC free article] [PubMed] [Google Scholar]
30. Mirimanoff RO, Gorlia T, Mason W, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol. 2006;24:2563–2569. [PubMed] [Google Scholar]
31. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. [PubMed] [Google Scholar]
32. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. [PMC free article] [PubMed] [Google Scholar]
33. Beetz C, Bergner S, Brodoehl S, et al. Outcome-based profiling of astrocytic tumours identifies prognostic gene expression signatures which link molecular and morphology-based pathology. Int J Oncol. 2006;29:1183–1191. [PubMed] [Google Scholar]
34. Li A, Walling J, Ahn S, et al. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res. 2009;69:2091–2099. [PMC free article] [PubMed] [Google Scholar]
35. Mischel PS, Shai R, Shi T, et al. Identification of molecular subtypes of glioblastoma by gene expression profiling. Oncogene. 2003;22:2361–2373. [PubMed] [Google Scholar]
36. Nutt CL, Mani DR, Betensky RA, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63:1602–1607. [PubMed] [Google Scholar]
37. Shai R, Shi T, Kremen TJ, et al. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene. 2003;22:4918–4923. [PubMed] [Google Scholar]
38. Shirahata M, Iwao-Koizumi K, Saito S, et al. Gene expression-based molecular diagnostic system for malignant gliomas is superior to histological diagnosis. Clin Cancer Res. 2007;13:7341–7356. [PubMed] [Google Scholar]
39. Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin Cancer Res. 2004;10:5316–5326. [PubMed] [Google Scholar]
OTHER FORMATS
PubReader PDF (1.7M)
ACTIONS
Cite
Favorites
SHARE
RESOURCES
Similar articles
Cited by other articles
Links to NCBI Databases
FOLLOW NCBI
Connect with NLM
National Library of Medicine
8600 Rockville Pike
Bethesda, MD 20894
Web Policies
jon_k84
Re: Johnni post# 539267
Saturday, November 26, 2022 10:41:42 PM
Post#
539270
of 539351
There are multiple 10 year survivors in the DCVax trial. The actual doctors that participated in the study have confirmed this. I don’t know exactly how many, but I remember a bare minimum of 5 individuals that have personally expressed this testimony on social media. I would wager it’s around ~20 people if I had to guess.
And by the way, this is utterly unheard of in GBM
dstock07734
Member Level
Re: jon_k84 post# 539175
Saturday, November 26, 2022 4:50:45 PM
Post#
539180
of 539351
Yes. LP had to tread carefully. Look what happened to Dendreon which submitted BLA in 2006 and the approval was granted in 2010.
https://www.drugs.com/history/provenge.html
LP certainly doesn't want the same thing happened to NWBO. The way I see it from the approval history of keytruda is that once DcVax gets approval, for other indicators the approval process will accelerate. Of course, this is only doable when NWBO partners with some BPs.
https://www.drugs.com/history/keytruda.html
The first one is for Newly diagnosed GBM, the second is for Recurrent GBM. So, there are two different trials?
Amazing saga. This will make quite the movie. Ex, do you know any of these people? I believe you’ve had an interest in DC science for quite some time.
flipper44,
That is correct as usual and as you may remember there was then later on an issue between Cenkos, where NWBO director Dr. Navid Malik worked and connected with Mr. Neil Woodford, and Navid that ended up forcing him out of his job due to work conditions. Later on then after Mr. Cofer Black joined NWBO the then CEO of Cenkos Jim Durkin announced “retirement” without warning leaving Cenkos to scramble for a new CEO though Mr. Durkin agreed to stay until that was completed. Nothing normal about that exit strategy but of course if tied to Ondra and Mr. Woodford and what happened with NWBO Mr. Black May have offered that as the only acceptable choice to all parties without going to court and creating discovery issues for a major firm in the UK through which NWBO was seeking favor for approval. The potential for this scenario is there anyway.
All this to say that Linda’s only truly credited price “surprise” might see a kind of repeat with similar repercussions. She has certainly set the historical table for this quite well. Best wishes.
@ATLnsider
In Dr. Liau May 2021 presentation, around 46:45 minute mark she says UCLA is collaborating with Bristol Myers Squibb (BMS) $BMY on #DCVax-L + PD-1 inhibitor + CSF-1 inhibitor combo trial.This is good for $NWBO & may mean they will partner with $BMY & $MRK:
youtube.com
Immunotherapy for Glioblastoma: Overcoming Resistance with Linda M....
Dr. Linda Liau, Professor and Chair of the Department of Neurosurgery at UCLA, presents at the Ved P. Sachdev, MD Memorial Lecture at the 2021 Mount Sinai Ne...
2:25 AM · Nov 27, 2022
·Twitter for iPhone
That BK scare makes you vote yes? Clearly LP holding your vote hostage that she’ll bk the company if she don’t get all yeses? LP can announce an emergency meeting so we get majority of the vote after failing to get it from ASM but with a plan in favor for shareholders and any without a quiet period BS or start selling the c shares to get funds or maybe start selling the company to BP. She can keep the lights on without AS increase cut the BK BS.
Approval will come in time without a doubt. DCVax will be the next platform and yes Linda will get my yes vote.
That's simple. Because it is Merck's best interest to keep flying under the radar.
who's "we"? i seem to remember a stream of much larger shareholders calmly explaining they were voting yes, after you assumed your opinion represented majority sentiment.
explain to me your strategy for voting no? you are going to call the manager on our leadership and voice outrage that the PPS is not higher?
i'll be voting yes because, as a shareholder, i don't want to bankrupt the company on the verge of finalizing success.
Ex,
Ok I’ll bite on the fact that the trial endpoints were changed late in the game.
Now let’s go to the paper you cite about changing trial endpoints. In that paper they rightly reference the fact that changes to endpoints can lead to bias in the data
Please explain how changing the endpoints biased the OS data? How did the Statistical Plan permit patients to live longer?
Will MIA approval be sufficient to get our vote?
Will MIA approval and MAA submission announcement be sufficient to get our vote?
When I see articles suggesting new trials should be done my first reaction is “go and explain that to newly diagnosed GBM”.
They have no options. With Dcvax, they can hope to survive.
Thanks for sharing.
Thanks, Bright Boy!
Hi Gary, I agree with everything you’ve said and I think part of the reason management delayed the publication of the JAMA article for 3 months is because they were waiting on manufacturing approval at Sawston and Flaskworks approval. I bet we see these approvals soon!
I'm not suggesting that a potential partner wouldn't take preferreds, but only if they know the company has sufficient shares authorized to convert them.
is it really so hard to keep loose track of public presentations, with videos and slides, given by one of our PIs?
watching youtube videos with labeled chapter headings isn't exactly reading tea leaves.
(whoops, meant to reply to post 539321)
ex you are very talented at scrapbooking! have you considered reducing your carbon footprint and just doing some of these posts with scissors and glue? perhaps you could write a letter to the JAMA editors composed of letters cut out of magazines about all the worrying aspects of this trial.
i hope you're coping with the fact that actual experts in the field share none of your concerns. be well!
Thanks for posting this.
I sent this email to them per their request at the end of the article:
Dear Ms Houser,
There are important allegations in your article that are wrong.
Namely when you state:
"When the terms of an analysis are defined after results have already been seen, it’s called a “post-hoc analysis,” and it’s a controversial way to assess a treatment’s efficacy. “The more analyses that are done post-hoc, the greater chance of finding results by chance — meaning that they are not true results"
Post-hoc analysis is defined as: "Post hoc in Latin means 'after this'. Simply put, a post-hoc analysis refers to a statistical analysis specified after a study has been concluded and the data collected. A post-hoc test is done to identify exactly which groups differ from each other. Therefore, such tests are also called multiple comparison tests." Post-hoc analysis is also at times called Data dredging -- sometimes referred to as data fishing -- is a data mining practice in which large data volumes are analyzed to find any possible relationships between the data. Data scientists can then form hypotheses about why these relationships exist. That did not happen here.
The analysis for the DCVAX-L Phase-3 trial was not "post-hoc" because the SAP (Statistical Analysis Plan) was updated and submitted before the trial data was unblinded. Updating the SAP is common before unblinding as stated here: "The plan should be reviewed and possibly updated as a result of the blind review of the data and should be finalized before breaking the blind." http://onbiostatistics.blogspot.com/2020/01/pre-specification-and-statistical.html
If you do you Due Diligence you will find your allegation that the SAP was done after data lock is completely incorrect If you review the their October 2020 NWBO press release:
https://nwbio.com/northwest-biotherapeutics-announces-data-lock-of-phase-iii-trial/
Northwest Biotherapeutics Announces Data Lock of Phase III Trial
05 OCT 2020
FOR IMMEDIATE RELEASE
CONTACTS
Dave Innes
804-513-4758
dinnes@nwbio.com
Les Goldman
240-234-0059
lgoldman@nwbio.com
BETHESDA, Md., October 5, 2020 – Northwest Biotherapeutics (OTCQB: NWBO) (“NW Bio”), a biotechnology company developing DCVax® personalized immune therapies for solid tumor cancers, today announced that the database for the Phase III trial of DCVax®-L for Gliobastoma has been locked.
With the database now locked, the independent service firms managing the Clinical Trial are arranging for the independent statisticians to have access to the unblinded raw data from the Trial. Neither the Company nor any party other than the independent statisticians will have access to any unblinded data at this stage.
The statisticians will proceed as quickly as possible with analyses of the raw data and prepare summaries of the Trial results for review by the Company, the Principal Investigator, the Steering Committee of the Trial, the Scientific Advisory Board, and a panel of independent brain cancer experts, who will analyze the data with the statisticians in preparation for public announcement and scientific publication.
“We are excited to be so close to the finish line now, after such a long road” commented Linda Powers, the Company’s CEO. “We are hopeful that DCVax®-L can become an important new treatment option for patients who urgently need more and better treatments for Glioblastoma brain cancer.”
“We are grateful to the independent service firms and the clinical trial sites who have worked so hard to complete the data collection and confirmation during many months of COVID restrictions and challenges,” Ms. Powers continued. “We are also very grateful to our shareholders for their patience and support, which has made all this possible.”
How could the statisticians proceed with analyses of the raw data and prepare summaries of the Trial results without the final SAP?
They couldn't.
. . . and the Principal Investigator, the Steering Committee of the Trial, the Scientific Advisory Board, and a panel of independent brain cancer experts, who analyze the data with the statisticians in preparation for public announcement and scientific publication would not let them change SAP after all this is done . . . and even if NWBO tried to do that do you think they would remain silent. These panels, boards and committees are put in place to ensure nothing untort/nefarious like data dredging happens. That could happen for even a microsecond. That is why these people were put in place.
I hope you find my comments and feedback helpful
Resus, No stepping on this egg! I think we’ll be happy with what happens these next few weeks! Or days…
+++ #DDAmanda Chart on: $NWBO (With Notes):
DDAmanda lets you scan for these b4 they run:

#DDAmanda Christmas Special: https://ddamanda.com/SignUpXMAS.php
#DDAmanda Notes (Every stock has notes. SS Changes are also tracked):
7-5-22 - OS Inc - New: 1033, Old: 1021, Chg: +12, +1% **
5-24-22 - FL Inc - New: 918, Old: 860, Chg: +58, +7% **
8K: 11-29-21 - material definitive agreement **
11-24-21 - Verified Chg - New: Verified **
11-24-21 - FL Inc - New: 860, Old: 658, Chg: +202, +31% **
11-18-21 - Verified Chg - New: Verified **
10-21-21 - OS Inc - New: 921, Old: 899, Chg: +22, +2% **
8K: 9-1-20 - material definitive agreement **
8K: 8-20-20 - material definitive agreement **
8K: 5-27-20 - material definitive agreement **
8K: 4-1-20 - material definitive agreement **
8K: 5-28-19 - material definitive agreement **
8K: 12-11-18 - material definitive agreement **
8K: 11-13-18 - material definitive agreement **
8K: 6-28-18 - securities purchase agreement **
8K: 5-7-18 - material definitive agreement **
Def14: 4-12-18 - increase the number of authorized **
Def14: 4-9-18 - increase the number of authorized **
Pre14: 4-4-18 - increase the number of authorized **
Pre14: 3-26-18 - increase the number of authorized **
8K: 3-20-18 - material definitive agreement **
8K: 1-4-18 - material definitive agreement **
8K: 12-7-17 - material definitive agreement **
8K: 11-21-17 - lawsuit **
8K: 10-16-17 - material definitive agreement **
8K: 9-22-17 - material definitive agreement **
8K: 8-7-17 - material definitive agreement **
8K: 6-28-17 - material definitive agreement **
8K: 6-13-17 - material definitive agreement **
8K: 5-31-17 - material definitive agreement **
8K: 5-26-17 - repurchase **
8K: 4-25-17 - material definitive agreement **
8K: 3-23-17 - material definitive agreement **
8K: 3-23-17 - material definitive agreement **
8K: 3-10-17 - material definitive agreement **
8K: 3-7-17 - material definitive agreement **
8K: 1-19-17 - 100% of the **
8K: 12-23-16 - repurchase **
8K: 12-9-16 - notice of delisting **
8K: 10-19-16 - material definitive agreement **
8K: 9-15-16 - material definitive agreement **
8K: 9-9-16 - material definitive agreement **
8K: 9-6-16 - material definitive agreement **
8K: 8-23-16 - material definitive agreement **
8K: 7-11-16 - securities purchase agreement **
8K: 6-30-16 - reverse stock split **
8K: 5-16-16 - material definitive agreement **
8K: 5-2-16 - notice of delisting **
Was NWBOD 10-23-12: **
Was NWBO 09-25-12: 1-16 R/S **
Jo**R 7-19-19 (1) Massive Dilution looks like.
Z
"Good things take time, but great things happen all at once."
We may or may not be heading into one of those great times. We shall see.
BMS and MRK. Why isn’t news like
This ever releases. Why is it we always have to dig it up.
It's a vote to re-elect Ms. Powers and Dr. Malik:
"The persons named in the enclosed proxy will vote to elect each of Ms. Powers and Dr. Malik as a Class III director unless your proxy is marked otherwise."
I agree with you ATL that the collaboration would be with both BMS and Merck. I do not know if combination trial with BMS has started yet or not? Please someone confirm this.
They have said many times that they have long term survival in combination trial. So, based on this I think they are far into program collaborating with Merck and the data hopefully would unbounded next year in first half. Keep in mind we are talking about rGBM here which the survival rate known to be much less than 12 months. So, we should be able to get the data very quickly. If 50% of patient living longer than 12 months that destine for approval no matter what.
In addition, people talking about the termination of trial with BMS back in 2020. If you listen carefully what Dr. LL said earlier, she mentioned that the timing of administering of the drug specially from BMS had different results. I think after that study they came back with the new one based on what they have leaned earlier and collaborating with BMS now.
I'd say that this terminated trial is a different one as there's no mention of the CSF-1 receptor inhibitor, only DCVax+Nivolumab.
Also that trial was terminated at latest on July 24, 2020.
LL's presentations including the BMS collaboration trial slides were held in 2021 and 2022.
Interesting:
I am not familiar with the US corporate law but the first purpose of ASM is about adding two members to $NWBO Board of Directors to serve as Class III Directors for a term of three years. Could be for someone from $BMY & $MRK 🤔#partnership 🥂🌏
— voloda (@voloda37814097) November 27, 2022
Thanks for sharing, good observation.
In Dr. Liau May 2021 presentation, around 46:45 minute mark she says UCLA is collaborating with Bristol Myers Squibb (BMS) $BMY on #DCVax-L + PD-1 inhibitor + CSF-1 inhibitor combo trial.This is good for $NWBO & may mean they will partner with $BMY & $MRK: https://t.co/WeCYXqysU5
— ATLnsider (@ATLnsider) November 27, 2022
Yes, I was not saying it wasn’t, only that it is a TLR3 Agonist. You asked if there were other suppliers of Poly ICLC, and I confirmed that is the generic name for Hiltonol, which is the brand name and it comes from Oncovir. But there are other TLR3 Agonists made by other parties and they all have patents for their way of making it, just like there are a variety of PD-L1 and PD1 blockers, made by different means. And there are likely now patents that have expired, since it has been around for 40 years. Not sure if they are all exactly the same in efficacy, as some are more water soluble. But the point further was that the drug is being studied in many places and has not proven to be efficacious so far by itself and that any manufacturer would probably love to work together. They also are using it in many vaccine applications and not just cancer, so it’s not like they have an incentive to exclude DCVax-L from having access or doing a deal with them.
There was a previous trial for BMY, but it did not go forward and MRK stepped in directly with UCLA.
I think most have thought the two were jockeying. But since the Opdivo/Nivolumab trial with DCVax-L did not start, it appeared BMY was sidelined. The contract was pulled and then the Keytruda trial started instead.
BMY Opdivo/Nivolumab trial:
https://clinicaltrials.gov/ct2/show/NCT03014804
I agree it's BMY vs MRK given the UCLA combination trials.
Hey Bio,
Could it be that the Poly-ICLC used in this DCVax+Keytruda+Poly-ICLC combo trial ( https://clinicaltrials.gov/ct2/show/NCT04201873 ) actually is Hiltonol as under the 'Intervention/treatment' section it says;
Drug: Poly ICLC
Given IM
Other Names:
Hiltonol
Poly I:Poly C with Poly-L-Lysine Stabilizer
poly-ICLC
PolyI:PolyC with Poly-L-Lysine Stabilizer
Polyinosinic-Polycytidylic Acid Stabilized with Polylysine and Carboxymethylcellulose
Polyriboinosinic-Polyribocytidylic Acid-Polylysine Carboxymethylcellulose
Stabilized Polyriboinosinic/Polyribocytidylic Acid
Maybe not any significance in that even if it really is Hiltonol. But anyways I found it even somewhat interesting bit
Excellent! Some peeps still clueless
Thanks Doc logic, I wanted the video watcher to listen to the start of the conversation and questions about the combo trial, so I recommended starting at the 46:45 minute mark to have more context.
ATLnsider,
That mention occurred about at the 48 and change mark after the question about macrophages. Oh by the way, have you heard my challenge to the bears over the years about what NWBO knows about macrophages and why THAT is one of the key foundations to the scientific rational for Direct?; ). Dr. Linda Liau explains why, in the less aggressive form of DCVax known as L, that there is a strong rational for introducing CSF-R1 inhibitor. Now where do you think she got THAT idea from?; ). Maybe she has been made aware of how Direct functions to some degree right? This is absolutely so fun to watch at this stage and yes BMY is mentioned as the collaborator for a future trial which I take to mean is they have been introduced to keep Merck shareholders aware of what is to gain and what is to lose when a bid is made to partner. Linda Powers has taught Dr. Linda Liau about leverage it seems. What a team!; ). Best wishes.
Assume you are talking about this trial.
https://clinicaltrials.gov/ct2/show/NCT04201873
It is possible that we can expect the data early next year. Linda Liau mentioned nine months ago in the presentation that 20 patients had been recruited last year. The rest 20 can be recruited in a year or two. We can assume by the end of this year, the recruitment is done.
There are two combinations in the trial if I am not mistaken.
Combo 1
DcVax + poly-iclc
Combo 2
DcVax + poly-iclc + keytruda
I do think it is possible that UCLA is collaborating with both BMS (BMY) & Merck (MRK).
It is not self-administered. The YouTuber made a mistake about that.
Thank you Larppis, for confirming that Dr. Liau did indeed say that UCLA is collaborating with BMS on the combo trial with: DCVax-L + PD-1 inhibitor + CSF-1 receptor inhibitor.
I do think it is possible that UCLA is collaborating with both BMS (BMY) & Merck (MRK).
How is the vaccine self administered after the initial batch?
Gary
Mr. Bigger seems to expect data on the Keytruda trial at the beginning of next year:
T+3 years not days. T early 2020
— Michael Bigger (@biggercapital) November 27, 2022
|
Followers
|
1649
|
Posters
|
|
|
Posts (Today)
|
185
|
Posts (Total)
|
726693
|
|
Created
|
02/02/05
|
Type
|
Free
|
| Moderators flipper44 sentiment_stocks CaptainObvious Poor Man - Doc logic JerryCampbell | |||
![]()
“Now this is not the end. It is not even the beginning of the end. But it is, perhaps, the end of the beginning.”
~ Winston Churchill
Stylized Dendritic Cell featured on NWBO board since 2015

- Dr. Linda Liau, PhD, MBA, Professor and Chair, Department of Neurosurgery, David Geffen School of Medicine at UCLA
Clinical Trials
DCVax®-L to Treat Newly Diagnosed GBM Brain Cancer (NCT00045968) - Phase III (Double Blind)
UK (MHRA): DCVax-L to Treat Newly Diagnosed GBM Brain Cancer (EudraCT#) 2011-001977-13
DE (Germany - PEI): DCVax-L to Treat Newly Diagnosed GBM Brain Cancer (EudraCT#) 2011-001977-13
Expanded Access Protocol for GBM Patients with Already Manufactured DCVax®-L Who Have Screen-Failed Protocol 020221 (NCT02146066) (Expanded Access)
Safety and Efficacy Study of DCVax-Direct in Solid Tumors (NCT01882946) - Phase I/Phase II (Open Label)
UK Clinical Trials - Study of a Drug (DCVax®-L) to Treat Newly Diagnosed GBM Brain Cancer
EU Clinical Trials for DCVax-L - Phase III
Dendritic Cell Vaccine for Patients with Brain Tumors (NCT01204684) - Phase II - at UCLA - Randomized (Open Label) testing DCVaccine with Resiquimod and DC Vaccination with Adjuvant polyICLC
Pembrolizumab and a Vaccine (ATL-DC) for the treatment of Surgically Accessible Recurrent Glioblastoma - Phase 1 (NCT04201873)
Dendritic Cell-Autologous Lung Tumor Vaccine (DCVax-L) and Nivolumab in Treating Patients with Recurrent Glioblastoma - Phase 2 (NCT03014804)
Dendritic Cell Therapy for Brain Metastases From Breast or Lung Cancer (NCT0368765) - Phase 1 - Collaborator: Mayo Clinic
Announcement of DCVax-L and Anti-PD-1 Monoclonal Antibody (Pembrolizumab) for Patients with Liver Metastases of Primary Colorectal Carcinoma Phase 2 Trial - November 17, 2016 - University Medical Center (UMC) of the Johannes Gutenberg University of Mainz
Cognate Bioservices - Owned by Charles River Labs
Website
Company Contact Info
Investor Relations:
Les Goldman (Company) (202) 841-7909 lgoldman@nwbio.com
Sign up for Northwest email list here (hit the subscribe to email list button in the lower right)
Company Headquarters
4800 Montgomery Lane, Suite 800, Bethesda, MD 20814 (240) 497-9024
NW Bio is developing cancer vaccines designed to treat a broad range of solid tumor cancers more effectively than current treatments, and without the side effects of chemotherapy drugs. NW Bio’s proprietary manufacturing technology enables them to produce its personalized vaccine in an efficient, cost-effective manner. NW Bio has a broad platform technology for DCVax dendritic cell-based vaccines.
Their lead product, DCVax-L, is currently in a 331-patient Phase III trial for patients with newly diagnosed Glioblastoma multiforme (GBM), the most aggressive and lethal brain cancer. This trial is currently underway at 69 locations thoughout the United States, Germany and the United Kingdom. NW Bio has also conducted a Phase I/II trial with DCVax-L for late stage ovarian cancer together with the University of Pennsylvania.
Their second product, DCVax-Direct, is currently in a 60-patient Phase I/II trial for direct injection into all types of inoperable solid tumor cancers, with trials currently being conducted at both MD Anderson Cancer Center in Texas, as well as Orlando Health in Florida.
They previously received clearance from the FDA for a 612-patient Phase III trial with its third product, DCVax-Prostate, for late stage prostate cancer.
DCVAX Survival Stories & Testimonials
Alice - Metastic Merkel Cell patient from Florida - ASCO 2018
Brad Silver - GBM patient from Huntington Beach, California - ASCO 2018
Sarah Rigby - GBM patient from Hong Kong - ASCO 2018
Kristyn Power - daughter of GBM patient from Canada - ASCO 2018
Kat Charles - GBM patients from UK - ASCO 2018 - as related by her husband Jason (Kat's Cure)
Prospective patients may contact NW Bio at patients@nwbio.com
UCLA Jamil Newirth DCVax-Patient Video - 2015
Allan Butler Video - National Geographic Vice President - DCVax-Direct patient from Phase 1 Trial with Pancreatic Cancer
NWBO - Patients Sunday Dennis and Jami Newirth - Enrolled at UCLA - Vimeo, Uploaded approx. May 2015
NWBO - Vaccine Helps Keep Brain Cancer Patient Alive (Jennifer Sugioka) - NBC Channel 4, Southern California, February 24, 2015
NWBO - National Geographic's Allan Butler Stage IV Pancreatic Patient using DCVax-Direct at MD Anderson
NWBO GBM Brain Cancer Survival Story of Mark Pace

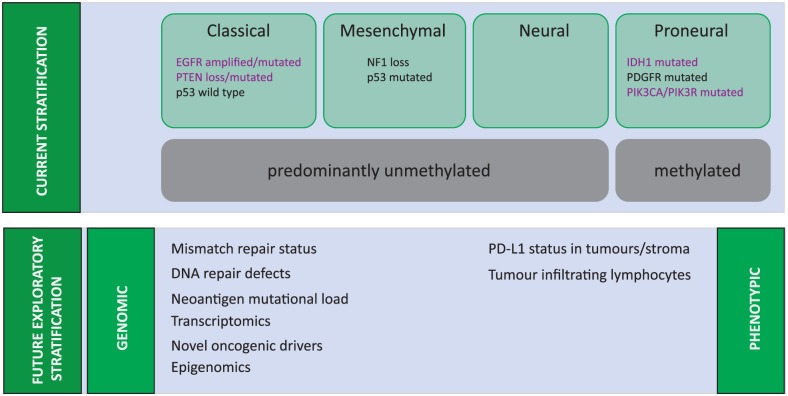
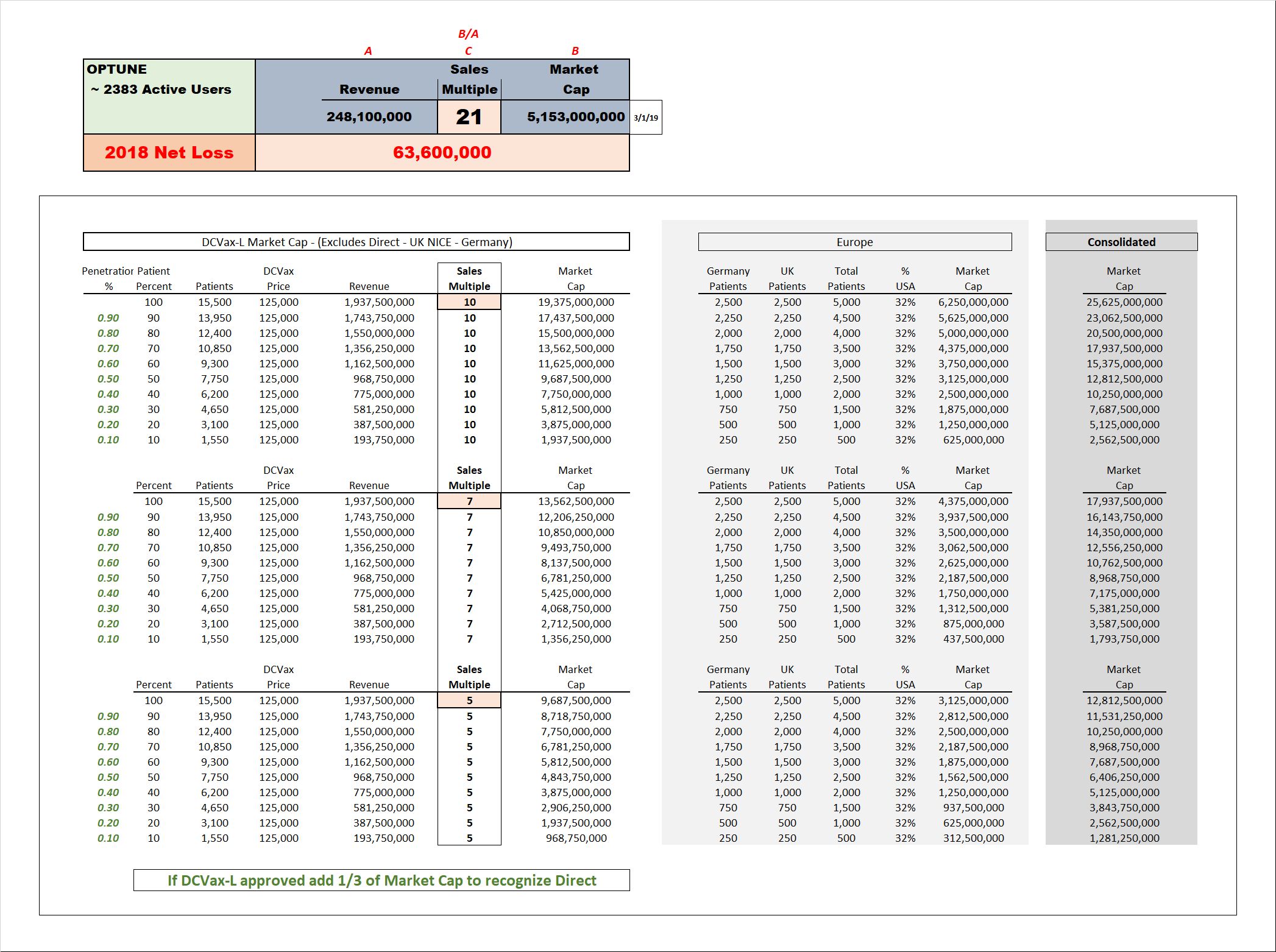
Presentations
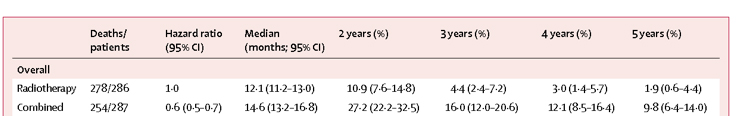
UCLA Agreements
Prostrate
DCVax-Phase II
DCVax-Booster
Upcoming Events
Videos
Linda M. Liau, MD, PhD, MBA - April 24, 2019 at University of Washington, Neurosciences Institute
| Volume | |
| Day Range: | |
| Bid Price | |
| Ask Price | |
| Last Trade Time: |



