 News
News  Market Data
Market Data  Discover
Discover 
Support: 888-992-3836
Copyright © 2023 InvestorsHub Inc.






genisi
![]()
Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
We can delete MDV-3100 now:
Medivation Gets Partner for Prostate Drug
http://www.thestreet.com/_yahoo/story/10617222/1/medivation-lands-partner-for-prostate-cancer-drug.html?cm_ven=YAHOO&cm_cat=FREE&cm_ite=NA
GSK/Genmab announced the accelerated approval of Arzerra (ofatumumab) from the FDA for use in patients with chronic lymphocytic leukemia (CLL) that is refractory to fludarabine and alemtuzumab.
http://www.reuters.com/article/marketsNews/idCNLR34044920091027?rpc=44
GAO finds FDA unwilling to pull drugs off the market, even when their benefits are unclear
http://www.sun-sentinel.com/features/health/sns-ap-us-fda-unproven-drugs,0,236194.story
MATTHEW PERRONE, October 26, 2009
WASHINGTON (AP) — The Food and Drug Administration has allowed drugs for cancer and other diseases to stay on the market even when follow-up studies showed they didn't extend patients' lives, say congressional investigators.
A report due out Monday from the Government Accountability Office also shows that the FDA has never pulled a drug off the market due to a lack of required follow-up about its actual benefits — even when such information is more than a decade overdue.
When pressed about that policy, agency officials said they have no plans to get more aggressive.
The GAO says the FDA should do more to track whether drugs approved based on preliminary results actually have lived up to their promise.
The FDA responded that the report paints an overly negative picture of its so-called "accelerated approval" program, which is only used to approve drugs for the most serious diseases.
"Millions of patients with serious or life-threatening illnesses have had earlier access to new safe and effective treatments," thanks to the program, the FDA said in its response to the report.
In 1992, the FDA began speeding up the approval of novel drugs based on so-called surrogate endpoints, or laboratory measures that suggest the drug will make real improvements in patient health. HIV drugs, for example, are cleared based on their virus-lowering power, a predictor of increased survival.
Drugmakers favor the program because it helps them get products to market sooner, without conducting long-term patient studies that can take years and cost hundreds of millions of dollars. A condition of quicker approvals is that drugmakers conduct follow-up studies to show the drug's benefits actually panned out.
But the GAO report, a copy of which was obtained by The Associated Press, identified several drugs still on the market that never lived up to their initial promise. And in the 16 years that the FDA has used accelerated approval, it has never once pulled a drug off the market due to missing or unimpressive follow-up data.
"FDA has fallen far short of where it should be for patient safety," said Sen. Charles Grassley, R-Iowa, who requested the investigation.
Of the 144 studies the FDA has required under the program since 1992, 64 percent have been completed and more than one-third are still pending, according to the GAO. Investigators said the FDA does not rigorously track whether companies are making progress on their required studies, although the agency is improving.
FDA officials say they have overhauled their tracking system since the GAO completed its report. And an outside analysis by contractor Booz Allen Hamilton concluded last month that most companies are meeting their study requirements on time.
But in the case of Shire Laboratories' low blood pressure treatment ProAmatine, the required study has gone incomplete for more than 13 years. The GAO found that ProAmatine has generated more than $257 million in sales, even though "the clinical benefit of the drug has never been established."
Shire did not respond to a request for comment Friday.
In other cases, the FDA has failed to act even when company studies show drugs did not improve patient outcomes.
The FDA approved AstraZeneca's lung cancer drug Iressa in 2003 based on early results showing it reduced the size of tumors. But later studies showed the drug did not significantly extend patient lives.
The FDA has left the drug on the market, despite hundreds of reports of a sometimes fatal pneumonia.
FDA officials explain that access to Iressa has been restricted to a small number of patients who have shown benefit. The agency recommends all other patients try two alternative drugs.
Iressa "is not available to new patients," AstraZeneca confirmed in a statement.
The GAO concluded that the FDA has no policy for pulling drugs off the market that were approved using surrogate endpoints. When GAO investigators confronted FDA officials about this lack of enforcement, they reportedly said it would be "difficult, if not impossible," to draft a standard policy for withdrawals, given the unique circumstances of individual drugs.
In certain cases, FDA officials say withdrawing a drug would mean eliminating the only available treatment for a condition.
"FDA should explain the principles it uses to make decisions such as drug withdrawals," said Principal Deputy Commissioner Dr. Joshua Sharfstein, in an interview with the AP. "But we don't want to lock ourselves into a specific set of criteria that takes away the flexibility to do what's right for the public health."
Sharfstein added that the agency has a task force assigned to look at policies like drug withdrawals.
Some consumers advocates say that's not good enough.
"The FDA has talked a lot about doing more enforcement, but this is an area where they're basically defending not enforcing the law," said Dr. Sidney Wolf, of the consumer advocacy group Public Citizen.
Wolfe said the lax policy sends a message to companies that there is no penalty for failing to complete studies.
The GAO recommends the FDA clarify when it will pull drugs off the market.
"As the scientific experts charged with overseeing the use of drugs it approves, FDA should be in a position to implement this recommendation," the report states.
BioCryst is seeking approval for the intravenous version of the drug through a pre-emergency use authorization, which would allow the company to ship the drug to the government for one year as a preventive measure to stop a large flu outbreak.
Thanks, that's what I meant.
Kamada Ltd. (TASE: KMDA) raised NIS 95 million (~$26M) in an issue of convertible bonds and NIS 35 million in a rights issue.
Kamada just payed off its $20M debt to Hercules Technology Growth Capital Inc. (Nasdaq: HTGC) using some of this money. The rest will be used to finance the inhaled AAT program.
At this early stage, and after only reading the short eurekalert summary and not the American Journal of Human Genetics manuscript, I'll stick to "interesting" :)
Agree on PTC - intriguing and worth watching.
Re: NVS vs Teva Lotrel litigation
Teva launched and sells generic lower dosages Lotrel and there isn't a date for the patent trial yet. NVS launched its generic and settled with Lupin and Par. So I believe only the lower doses are available as generics.
Interesting, thanks for posting.
Did AMGN say if D-Mab resubmission is going to be class I or II?
US advisers decline to push Gardasil for boys
http://www.reuters.com/article/rbssHealthcareNews/idUSN2147912020091021
* Committee advises that doctors free to use vaccine
* Says needs more evidence of cost benefit
* Did not consider value in preventing cancer (Updates throughout, adds quotes, share price)
By Maggie Fox, Health and Science Editor
WASHINGTON, Oct 21 (Reuters) - U.S. vaccine advisers declined to press for the use of Merck & Co's (MRK.N: Quote, Profile, Research, Stock Buzz) Gardasil in boys and men, opting instead on Wednesday to simply advise doctors they are free to use it.
Despite some impassioned pleas from patients and doctors alike, the Advisory Committee on Immunization Practices voted almost unanimously for "permissive" use of the vaccine for boys. It protects against the human papillomavirus, or HPV, which causes a variety of cancers and genital warts.
But the committee did recommend including Gardasil for eligible boys aged 9 to 18 in the Vaccines For Children program, a government-funded system that provides vaccines to children eligible for the state-federal Medicaid health insurance plan and other uninsured children.
Merck's shares were down 1.7 percent at $33.16 in afternoon trading on the New York Stock Exchange.
The U.S. Centers for Disease Control and Prevention currently recommends Gardasil for girls 11 and 12 years old and women 13 to 26 who have not been vaccinated.
Earlier this month, the U.S. Food and Drug Administration approved Gardasil for preventing genital warts in boys and men ages 9 through 26.
The main reason the vaccine was approved was to prevent cervical cancer, which kills 4,000 women a year in the United States alone. But various strains of HPV also cause disfiguring genital warts, anal and penile cancers and head and neck cancers.
"We know that the later the cancer is discovered, the lower the chance of survival is," David Hastings of the Oral Cancer Foundation told the committee, asking for a recommendation to add the vaccine to the standard schedule for boys.
However, ACIP decided only to consider its use based on its ability to prevent genital warts. There, the costs did not seem to justify using a $360 vaccine.
"They are sending a, very unfortunately in my opinion, weak message to physicians," said Dr. William Schaffner of Vanderbilt University in Tennessee and member of the National Foundation for Infectious Diseases.
But Dr. Diane Solomon of the National Cancer Institute said ACIP had an obligation for "responsible stewardship" of taxpayer money. "I think it is important to communicate to industry that we are not going to blithely indicate excessive costs," Solomon said.
Gardasil, sold as a series of three vaccines, had global sales of $1.4 billion in 2008, with an additional $865 million received through a joint venture with Sanofi-Aventis SA (SASY.PA: Quote, Profile, Research, Stock Buzz).
Earlier on Wednesday, the ACIP voted to recommend the use of GlaxoSmithKline Plc's (GSK.L: Quote, Profile, Research, Stock Buzz) rival Cervarix vaccine for routine administration among girls 11 and 12 years old.
ACIP members said they doubted many insurers would cover HPV vaccines for boys.
Merck said it was planning a rebate program before the end of the year. "The rebate program for Gardasil enables eligible privately insured 19-26 year olds whose out-of-pocket costs are over $30 to receive a rebate from Merck for up to a maximum of $130 per dose," the company said in a statement.
Thanks, Roy. Nice to see someone actually bothered opening the link :)
This was expected:
http://siliconinvestor.advfn.com/readmsg.aspx?msgid=25913930
TEVA has filed an ANDA for generic Nuvigil:
http://finance.yahoo.com/news/Cephalon-Notified-of-Generic-prnews-2211795615.html?x=0&.v=1
First Nuvigil ANDA
Cephalon Notified of Generic Filing for Armodafinil Tablets
http://finance.yahoo.com/news/Cephalon-Notified-of-Generic-prnews-2211795615.html?x=0&.v=1
FRAZER, Pa., Oct. 21 /PRNewswire-FirstCall/ -- Cephalon, Inc. (Nasdaq: CEPH - News) today announced receipt of a Paragraph IV Certification Notice Letter on October 20, 2009 regarding an Abbreviated New Drug Application (ANDA) submitted to the U.S. Food and Drug Administration (FDA) by Teva Pharmaceuticals USA, Inc. requesting approval to market and sell a generic version of the 50 mg, 100 mg, 150 mg, 200 mg and 250 mg strengths of NUVIGIL® (armodafinil) Tablets [C-IV]. In the Notice Letter, Teva alleges that U.S. Patent Nos. 7,132,570 (the "'570 Patent"), 7,297,346 (the "'346 Patent") and RE37,516 (the "'516 Patent") issued to Cephalon are invalid, unenforceable and/or will not be infringed by Teva's manufacture, use or sale of the product described in Teva's ANDA submission.
Cephalon is currently reviewing the Notice Letter. By statute, if Cephalon initiates a patent infringement lawsuit against Teva within 45 days of Cephalon's receipt of the Notice Letter, then the FDA would be automatically precluded from approving the Teva ANDA until the earlier of entry of a district court decision finding the patents invalid or not infringed or 30 months from the receipt of the Notice Letter by Cephalon.
Cephalon has a three-year period of marketing exclusivity for NUVIGIL that extends until June 15, 2010. In addition, including the six-month pediatric extension, the '516 Patent, the '346 Patent, and the '570 Patent expire on April 6, 2015, May 29, 2024, and June 18, 2024, respectively. Teva's Notice Letter does not challenge Orange Book-listed U.S. Patent No. 4,927,855 (the "'855 Patent"), which provides additional protection until October 22, 2010, the expiration date of the '855 Patent.
About Cephalon, Inc.
Lost in Transmission — FDA Drug Information That Never Reaches Clinicians
http://healthcarereform.nejm.org/?p=2126&query=home#
Lisa M. Schwartz, M.D., and Steven Woloshin, M.D., October 21st, 2009
The 2009 federal stimulus package included $1.1 billion to support comparative-effectiveness research about medical treatments. No money has been allocated — and relatively little would be needed — to disseminate existing but practically inaccessible information about the benefits and harms of prescription drugs. Much critical information that the Food and Drug Administration (FDA) has at the time of approval may fail to make its way into the drug label and relevant journal articles.
The most direct way that the FDA communicates the prescribing information that clinicians need is through the drug label. Labels, the package inserts that come with medications, are reprinted in the Physicians’ Desk Reference and excerpted in electronic references. To ensure that labels do not exaggerate benefits or play down harms, Congress might have required that the FDA or another disinterested party write them. But it did not. Drug labels are written by drug companies, then negotiated and approved by the FDA.
When companies apply for drug approval, they submit the results of preclinical studies and usually at least two phase 3 studies — randomized clinical trials in patients with a particular condition. FDA reviewers with clinical, epidemiologic, statistical, and pharmacologic expertise spend as long as a year evaluating the evidence. FDA review documents (posted at www.accessdata.fda.gov/scripts/cder/drugsatfda/) record the reasoning behind approval decisions. Unfortunately, review documents are lengthy, inconsistently organized, and weakly summarized. But they can be fascinating, providing a sense of how reviewers struggled to decide whether benefits exceed harms. Yet in many cases, information gets lost between FDA review and the approved label.
Sometimes what gets lost is data on harms. For example, in 2001, Zometa (zoledronic acid, Novartis) was approved for use in patients with hypercalcemia of malignancy. Approval was based on the results of two trials,1 in which 287 patients with cancer were randomly assigned to receive either 4-mg or 8-mg doses of Zometa or Aredia (pamidronate), the standard of care. According to the label, 8 mg of Zometa was no more effective than 4 mg in reducing calcium levels but had greater renal toxicity (see box on Zometa data). The numbers quantifying the renal-toxicity data for the 8-mg dose did not appear in the label, as they did for the 4-mg dose. But they did appear in the 98 pages of FDA medical and statistical reviews. Surprisingly, the reviews also noted that the 8-mg dose was associated with a higher rate of death from any cause than the 4-mg dose (P=0.03). These mortality data also did not appear in the label. Nor did they appear in the journal article reporting on these studies,2 which actually recommended the 8-mg dose for refractory cases. In 2008, the FDA approved an updated Zometa label with an explicit warning statement: “Renal toxicity may be greater in patients with renal impairment. Do not use doses greater than 4 mg.” Yet the mortality data are still missing from the label.

Sometimes, efficacy data get lost. Lunesta (eszopiclone) was approved in 2004 for chronic insomnia. Sepracor, its manufacturer, began an intense direct-to-consumer advertising campaign — spending more than $750,000 a day in 2007 — featuring a luna moth that transforms frustrated insomniacs into peaceful sleepers. Lunesta sales reached almost $800 million last year. Clinicians who are interested in the drug’s efficacy cannot find efficacy information in the label: it states only that Lunesta is superior to placebo (see box on Lunesta data).3 The FDA’s medical review provides efficacy data, albeit not until page 306 of the 403-page document. In the longest, largest phase 3 trial, patients in the Lunesta group reported falling asleep an average of 15 minutes faster and sleeping an average of 37 minutes longer than those in the placebo group. However, on average, Lunesta patients still met criteria for insomnia and reported no clinically meaningful improvement in next-day alertness or functioning.
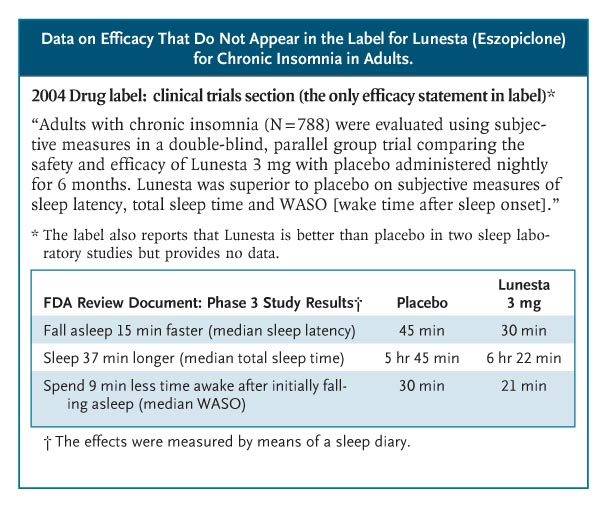
A sense of uncertainty about the net benefit of drugs is almost always lost. FDA approval does not mean that a drug works well; it means only that the agency deemed its benefits to outweigh its harms. This judgment can be difficult to make: benefits may be small, important harms may not have been ruled out, and the quality of the trials may be questionable. Since the nature — or even existence — of reviewer uncertainty is not addressed in the label, clinicians cannot distinguish drugs that reviewers endorsed enthusiastically from those they viewed with great skepticism.
Rozerem (ramelteon), for example, was approved in 2005 for chronic insomnia and was aggressively promoted to consumers. No efficacy data were provided in the label.4 The phase 3 sleep-laboratory studies that were included in the FDA’s medical review show that Rozerem reduced the time required for patients to fall asleep (as measured by polysomnography) by 14 minutes among younger adults and by 7 minutes among older adults (see box on Rozerem data). However, there were no subjective improvements in total sleep time, sleep quality, or the time it took to fall asleep. Two phase 3 outpatient trials confirmed that people didn’t notice much benefit from Rozerem. In a trial involving younger adults, Rozerem had no effect on any subjective sleep outcome; in one involving older adults, the drug reduced reported time to fall asleep by 7 minutes but did not reduce the proportion of cases meeting the definition of insomnia (taking more than 30 minutes to fall asleep). Nor did it improve any of the secondary outcomes: falling back asleep, number of awakenings, total sleep time, or sleep quality.
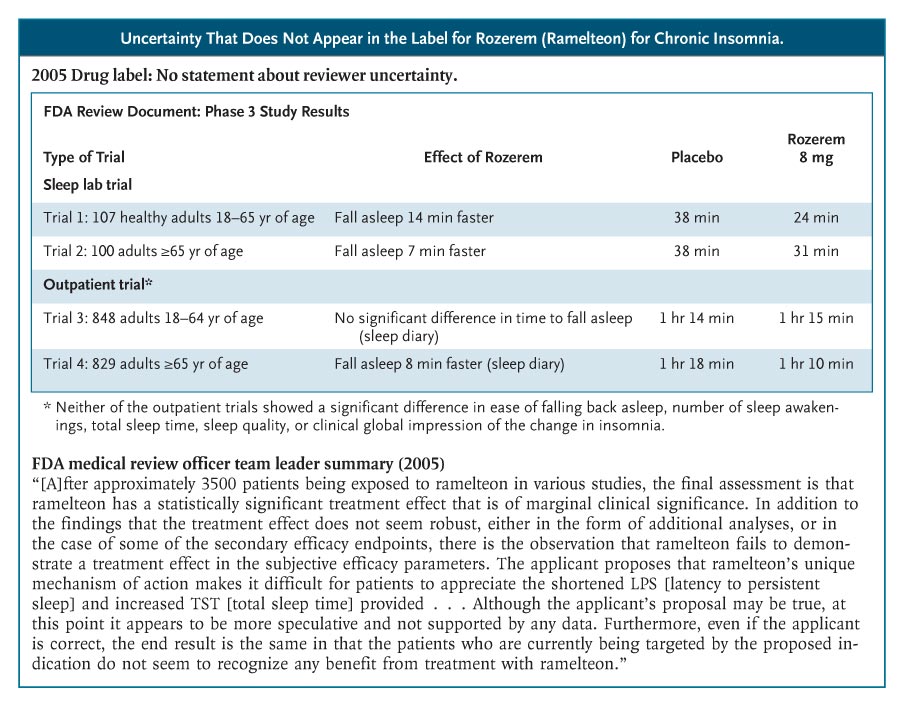
The Rozerem review included a memo from the medical review team’s leader, highlighting the team’s struggle to determine whether this drug provided any clinically important benefit and whether that benefit outweighed the harms. “Ordinarily,” the memo said, “a marginally clinically significant treatment effect would not preclude an approval of a product. However, the ability to approve such a product would then focus even more on the safety profile. . . . In the case of ramelteon, there are several issues in the safety profile of concern,” including frequent symptomatic side effects and possible hyperprolactinemia. The sense that the FDA’s decision was a close call was not communicated in the label.
To its credit, the FDA has recognized problems with drug labels. In 2006, it revised the label design, adding a “highlights” section to emphasize the drug’s indications and warnings. It also issued guidance about reporting trial results in the label, emphasizing the importance of effectiveness data. Yet the data presentations for the approval studies referred to in the labels for Lunesta and Rozerem, which were updated in 2009 and 2008, respectively, are substantively unchanged.
The FDA has not issued new guidance about its drug-review documents. A standardized executive summary of the reviews would be a substantial improvement. These summaries should include data tables of the main results of the phase 3 trials, highlight reviewers’ uncertainties, and note whether approval was conditional on a post-approval study.
Toward this goal, we conducted a pilot test, funded by the Robert Wood Johnson Foundation’s Pioneer Portfolio, in which FDA reviewers created “Prescription Drug Facts Boxes,”5 featuring a data table of benefits and harms. Recently, the FDA’s Risk Advisory Committee recommended that the FDA adopt these boxes as the standard for their communications. FDA leadership is deciding whether and how to use the boxes in reviews, labels, or both.
Whatever approach the agency adopts, it needs a better way of communicating drug information to clinicians. We don’t need to wait for new comparative-effectiveness results in order to improve practice. We need to better disseminate what is already known.
No potential conflict of interest relevant to this article was reported.
The views expressed in this article are those of the authors and do not necessarily reflect those of the Department of Veterans Affairs or the U.S. government.
Source Information
From the Dartmouth Institute for Health Policy and Clinical Practice, Hanover, NH; and the VA Outcomes Group, VA Medical Center, White River Junction, VT.
This article (10.1056/NEJMp0907708) was published on October 21, 2009, at NEJM.org.
References
1. Drugs@FDA. Approval history of NDA 021223: Zometa. Silver Spring, MD: Food and Drug Administration. (Accessed October 8, 2009, at http://www.accessdata.fda.gov/Scripts/cder/DrugsatFDA/index.cfm?fuseaction=Search.Label_ApprovalHistory#apphist.)
2. Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol 2001;19:558-567. [Free Full Text]
3. Drugs@FDA. Approval history of NDA 021476: Lunesta. Silver Spring, MD: Food and Drug Administration. (Accessed October 8, 2009, at http://www.accessdata.fda.gov/Scripts/cder/DrugsatFDA/index.cfm?fuseaction=Search.Label_ApprovalHistory#apphist.)
4. Drugs@FDA. Approval history of NDA 021782: Rozerem. Silver Spring, MD: Food and Drug Administration. (Accessed October 8, 2009, at http://www.accessdata.fda.gov/Scripts/cder/DrugsatFDA/index.cfm?fuseaction=Search.Label_ApprovalHistory#apphist.)
5. Schwartz LM, Woloshin S, Welch HG. Using a drug facts box to communicate drug benefits and harms: two randomized trials. Ann Intern Med 2009;150:516-527. [Free Full Text]
Sanofi-Aventis Inks $350M Deal for Wellstat’s Phase II Therapy for Type 2 Diabetes
[Wellstat Therapeutics is a privately-held biopharmaceutical company located in Gaithersburg, Maryland]
http://www.genengnews.com/news/bnitem.aspx?name=66031065&source=genwire
Sanofi-aventis negotiated an exclusive global license to Wellstat Therapeutics’ mid-stage type 2 diabetes therapy, PN2034, and related compounds. The deal could be worth up to $350 million to Wellstat in the form of an up-front cash payment plus development and regulatory milestones. Additional commercial milestones and royalties could follow.
PN2034 is an oral insulin sensitizer discovered by Wellstat. The drug’s ability to reverse insulin resistance in the liver of diabetic patients is currently being assessed in Phase II trials. PN2034 and its analogs reportedly act on a target that is distinct from other known types of insulin sensitizers. Wellstat says that PN2034 has also shown activity in models of fatty liver disease and other metabolic disorders.
Wellstat’s lead product is vistonuridine, an orally active prodrug of uridine. The drug has completed Phase III trials as an antidote to accidental overdose with the chemotherapy 5-fluorouracil (5-FU). Wellstat points out there are currently no licensed antidotes available for 5-FU overdose.
In May the company reported on 17 cases of 5-FU overdose for which its drug, vistonuridine, was requested and administered (within 8–96 hours of 5-FU overdose) under the FDA’s emergency-use IND provisions. Wellstat says that all 17 of the vistonuridine-treated patients recovered; a fatal outcome for at least 13 patients had been predicted due to the dose and rate of 5-FU administration.
Wellstat is separately investigating the potential use of vistonuridine as a combination therapy with 5-FU. A Phase III trial evaluating and comparing the treatment of pancreatic cancer using vistonuridine and high-dose 5-FU versus gemcitabine has been completed. Additional studies are being planned, including a Phase III trial in gastric cancer patients.
You were right on Replagal - "Shire announces plans to file a BLA with the FDA for REPLAGAL, its enzyme replacement therapy for Fabry disease, by the end of the year. The Company also announces that a treatment protocol for REPLAGAL, filed at the request of FDA, has been approved, and that it will support emergency IND requests. The early access program is being put in place in view of the announced supply restriction of the only currently marketed treatment for Fabry disease in the United States."
Updating the label with the increased PML risk with longer exposure, might bring more drug holidays.
Scientific Meeting Calendar
NOTE: ANYONE MAY UPDATE THIS FILE
Edits: Added entry for IDSA
OCTOBER 2009
American College of Rheumatology - ACR
October 17-21, 2009
Philadelphia, USA
http://www.rheumatology.org/annual/index.asp
Society for Neuroscience - SFN
October 17-21, 2009
Chicago, USA
http://www.sfn.org/am2009/
World Diabetes Congress - IDF
October 18-22, 2009
Montreal, Canada
http://www.worlddiabetescongress.org/
American Society of Human Genetics - ASHG,
October 20-24, 2009
Honolulu, Hawaii
http://www.ashg.org/2009meeting/
American College of Gastroenterology - ACG
October 23-28, 2009
San Diego, California
http://www.gi.org/physicians/education.asp
American Academy of Ophthalmology - AAO
October 24-27, 2009
San Francisco, CA
http://www.aao.org/meetings/annual_meeting/
The Obesity Society - Obesity 2009
October 24-28, 2009
Washington, DC
http://www.obesity.org/obesity2009/
Renal Week 2009
October 27 - November 1, 2009
San Diego, CA
http://asn-online.org/education%5Fand%5Fmeetings/renal%5Fweek/
Infectious Disease Society - IDSA
October 29 - November 1, 2009
Philadelphia, Pennsylvania
http://www.idsociety.org/IDSA2009.htm
American Association for the Study of Liver Diseases (AASLD)
Oct 31 - Nov 3, 2009
Boston, USA
http://www.aasld.org/thelivermeeting/Pages/default.aspx
American College of Chest Physicians - CHEST
October 31 - November 5, 2009
San Diego, California
http://www.chestnet.org/CHEST/program/about09.php
NOVEMBER 2009
American College of Allergy, Asthma & Immunology - ACAAI
November 5-11, 2009
Miami Beach, FL
http://www.acaai.org/Member/Annual_Meeting/Annual+Meeting.htm
American Heart Association - AHA
November 14-18, 2009
Orlando, Fl
http://scientificsessions.americanheart.org/portal/scientificsessions/ss/seeyounextyear2009
DECEMBER 2009
American Epilepsy Society - AES
December 4-8, 2009
Boston, MA
www.aesnet.org
American Society of Hematology - ASH
December 5-8, 2009
New Orleans, LA
http://www.hematology.org/meetings/2009/index.cfm
International Respiratory Congresses - AARC Convention
Dec. 5–8, 2009
San Antonio, Texas
http://www.aarc.org/education/meetings/#future_congress
American College of Neuropsychopharmacology - ACNP
Dec 6-10, 2009
Hollywood, Florida
www.acnp.org
--
Procedure for Updating Calendar
When adding or modifying entries, please follow these steps:
1. Copy the complete text from the old calendar.
2. Make your additions or modifications, inserting any new items in chronological order.
3. Near the top of the message, give a very brief description of your changes (e.g. “Edits: Added entry for AASLD”).
4. Post the updated calendar in a new message as a reply to the message with the old calendar.
BPAX/LibiGel more safety data in Phase III program
An independent data monitoring committee reviewed all unblinded adverse events in the safety study of LibiGel. There have been no deaths, one myocardial infarction and three breast cancers reported in the study. The committee recommended the continuation of the trial.
http://www.reuters.com/article/marketsNews/idCNBNG24088920091020?rpc=44
The Irish analysts were close with the numbers: Tysabri new weekly adds were about 230/week during Q3.
Glaxo kidney cancer drug - Votrient approved to treat patients with advanced renal cell carcinoma.
http://www.marketwatch.com/story/fda-approves-glaxosmithkline-renal-cancer-drug-2009-10-19?siteid=yhoof2
I'm quite certain Cervarix will be recommended for reimbursement for the prevention of cervical cancer in women, by ACIP but doubt the committee will find Gardasil for the prevention of genital warts in males, cost effective.
It's the 'safety first' era at the FDA.
Another one that got delayed by the FDA is ENDP's testosterone gel - fortesta:
http://www.reuters.com/article/marketsNews/idCNBNG50540420091019?rpc=44
US OKs Glaxo's cervical cancer shot
http://www.reuters.com/article/rbssHealthcareNews/idUSN1635263120091016
* Vaccine to be available later this year
* Glaxo's Cervarix competes with Merck's Gardasil
* Gardasil wins U.S. OK for males to prevent warts (Adds Merck, FDA comments)
By Lisa Richwine
LOS ANGELES, Oct 16 (Reuters) - GlaxoSmithKline Plc (GSK.L: Quote, Profile, Research, Stock Buzz) won U.S. approval on Friday to sell a cervical cancer vaccine for girls and young women, while a rival Merck & Co (MRK.N: Quote, Profile, Research, Stock Buzz) shot was cleared for preventing genital warts in males.
Glaxo said the U.S. Food and Drug Administration cleared the Cervarix vaccine for females ages 10 to 25.
Merck's competing cervical cancer vaccine, Gardasil, debuted on the U.S. market in 2006 and generated some controversy over its cost and potential side effects. On Friday, the FDA approved Gardasil for preventing genital warts in boys and men ages 9 through 26.
Both vaccines fight two strains of the sexually transmitted human papillomavirus (HPV) that cause about 70 percent of cervical cancers. Gardasil also targets two other HPV strains that cause genital warts. It is unclear how long protection lasts or whether booster shots will be needed.
Some analysts say it will be hard for Glaxo to compete against Merck's three-year head start in the U.S. market. Glaxo's original bid in 2007 stalled when the FDA sought more data.
Cervarix got a boost on Friday by winning the first approval for a cervical cancer vaccine in Japan, the world's second-largest pharmaceutical market after the United States.
Consensus forecasts suggest Cervarix will generate worldwide sales of just over $1 billion in 2013, according to Thomson Pharma, equivalent to about 2 percent of expected Glaxo sales for that year.
Merck's Gardasil had global sales of $1.4 billion in 2008. The company said Centers for Disease Control and Prevention advisory panel was expected to vote next Wednesday on whether to recommend Gardasil for males.
Both Cervarix and Gardasil fight two HPV strains that cause about 70 percent of cervical cancers.
In Glaxo's studies, Cervarix was 93 percent effective in preventing a pre-cancerous condition associated with the two strains. Cervarix also fought some other strains. It was 89 percent effective in preventing precancerous lesions from the third most common HPV type to cause cancer, Glaxo said.
Neither Gardasil nor Cervarix fights all types of HPV.
In the United States, cervical cancer is often caught early through Pap smears. Still, about 4,000 U.S. women die each year from the disease. Global deaths from the disease total about 280,000, mostly in developing countries where screening is lacking.
Some critics question if it is worth the cost to vaccinate for cervical cancer when its is largely treatable if caught early. Immunizing boys for genital warts also raised concerns since the warts often clear up without treatment.
Gardasil costs about $360, while Glaxo has not announced a price for Cervarix.
Glaxo said Cervarix should be available in the United States later this year. The most common adverse reactions in studies included injection site pain, fatigue and headaches.
FDA advisers in September said Cervarix appeared safe despite miscarriages reported around the time of vaccination and a small number of autoimmune problems such as rheumatoid arthritis and lupus. Panelists urged monitoring after approval to see if there was a link to the vaccine.
Cervarix contains a novel additive designed to boost the immune response. In a Glaxo study, Cervarix produced more antibodies than Gardasil but the researchers did not study if the higher levels provided more protection against cancer.
On Gardasil, the FDA said Merck will run post-approval studies to obtain more information about the safety and effectiveness in men and boys. The FDA has repeatedly said Gardasil remains safe with no unusual complications.
U.S. Seeks More Information on Novartis Lung Drug
http://abcnews.go.com/Health/wireStory?id=8859129
By Emma Thomasson
U.S. regulators have asked for more information on dosing proposed for a drug from Novartis AG for "smoker's lung," or chronic obstructive pulmonary disease, the Swiss group said on Monday.
Novartis said it would work with the Food and Drug Administration to review already submitted data for the drug, QAB149, as well as recently available data to determine what, if any, further clinical trials would be required.
"We think Novartis' attempts to build a respiratory franchise will only bear fruit once combination data and products are in the market, something that will not happen for many years," said Kepler Capital Markets analyst Tero Weckroth.
"From this point of view, a delay for QAB does not have a major impact on numbers," Weckroth said.
Novartis will initially sell QAB149 as a monotherapy. But industry analysts believe unlocking the medicine's full sales potential -- which some have put at $3-$5 billion a year -- will depend on combining QAB149 with other treatments.
Novartis is studying combinations with NVA237, a new drug it is developing with Vectura, and with Schering-Plough's inhaled steroid mometasone. Those combinations may not reach the market until around 2013, analysts estimate.
Novartis shares were trading flat at 51.95 Swiss francs by 3:38 a.m. EDT, versus a 0.5 percent rise in the DJ Stoxx European healthcare index. Vectura shares fell 3.2 percent.
QAB149, or indacaterol, is the first once-daily bronchodilator treatment for adult patients with COPD.
The European Medicines Agency recommended approval of the drug last month in a boost for Novartis' ambitions in respiratory medicine, a field currently dominated by GlaxoSmithKline, AstraZeneca and Pfizer.
"We are confident that Novartis will be able to address the FDA's dosing concerns over QAB149, particularly in view of the product's recent European approval," said PiperJaffray analyst Sam Fazeli.
Cytos anti-smoking vaccine missed target in mid-stage trial:
http://www.reuters.com/article/healthcareSector/idUSLG13562520091016
Teva currently holds about 11% in MediWound, which its leading product - Debrase has succeeded in phase III #msg-42629308
MediWound's Debrase hit two co-primary endpoints in its phase III trial and the trial will be stopped after treating the first 175 patients. Debrase will be marked by Teva and can be approved in Europe late next year.
I don't think Shire has capacity for more than 300 patients (US average dose) now, and it is likely to remain limited until its new manufacturing facility will fully function in 2012. Protalix said it will have enough capacity to supply 1,000 patients (US dosing) at launch and it can scale production more rapidly than Shire. However, I doubt Protalix will reach its capacity limit as its immediate market are the 400 Israelis patients.
Let's give chaperones a rest and wait for the data.
Enjoy your weekend.
Tarceva did not meet its secondary endpoint of OS in the ATLAS study (Avastin+/-Tarceva in maintenance setting in NSCL).
OSI Pharma posts higher Tarceva global sales in Q3
http://www.reuters.com/article/rbssHealthcareNews/idUSBNG43914120091015?sp=true
Says global sales rise 8 pct
* U.S. sales rise 7 pct
Oct 15 (Reuters) - OSI Pharmaceuticals Inc (OSIP.O) reported higher third-quarter global net sales for its cancer drug Tarceva, which it co-markets with Roche Holding AG (ROG.VX).
For the quarter ended Sept. 30, Tarceva's global net sales were about $301 million, of which U.S. accounted for about $118 million, the company said in a U.S. regulatory filing.
In the corresponding quarter last year, Tarceva's global net sales were $279 million, with U.S. contributing $110 million.
The company also said Roche informed that an exploratory data sweep for survival from the Atlas study on Tarceva was not positive.
In the trial, called Atlas, patients with advanced non-small cell lung cancer were treated with Tarceva in combination with Genentech Inc's Avastin. [ID:nN02472881]
Survival was a secondary endpoint in the Atlas study and the study was not powered to detect a significant difference in overall survival, OSI said.
OSI said it did not "opt-in" to the study because it was not designed to be registrational.
If the study was to result in a change to the Tarceva label, OSI would likely be required to opt-in and pay its retrospective share of the study costs and a penalty, it said.
OSI said it is not currently forecasting any opt-in payments for the study.
Hi M, PLX did say that full data will be presented at a medical conference. Meanwhile, as far as I can see the drug generated good data in the trial: both doses showed stat-sig reduction (change from baseline) in spleen vol at 9 months (p<.0001), and also hit this primary endpoint at 6 months. Same goes for 2 secondary endpoints - liver vol and hemoglobin levels. Platelet counts were stat-sig increased only with the higher dose but a trend was seen with the lower one. So bottom line on efficacy is it is not different from that of the other two competitors and patients will probably be dosed with 60U/kg (just like Cerezyme). No safety red lights so no worries here either.
Thanks for the congrats and hope that FOLD will live up to your expectations and not mine 
Trades up 15% at TASE on the news.
Protalix reports positive Phase III trial results
Oct. 15, 2009, 6:15 a.m. EDT LONDON (MarketWatch) -- Protalix BioTherapeutics on Thursday reported positive top-line results from its Phase III clinical trial of UPLYSO in treatment naive patients diagnosed with Gaucher disease. The company said the trial met its primary endpoint, mean reduction in spleen volume after nine months compared with baselines, at two different dosage levels. Statistically significant improvements compared with baselines were also observed in the secondary endpoints. The company said it plans to present more comprehensive results in the near future.
Wyeth will transfer Relistor back to Progenics along with $10M within a year and Progenics immediately take over development of an oral formulation.
http://www.reuters.com/article/americasRegulatoryNews/idUSN1320573420091014
Maybe if you switch the word 'bad' to 'no' :)
Any MS patients speak or present on the panel?
Jim, what happened between #msg-42453668 and today's one on ACOR?

