News
News  Market Data
Market Data  Discover
Discover 
Support: 888-992-3836
Copyright © 2023 InvestorsHub Inc.


Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
Lipocine Announces Positive LPCN 2401 Clinical Results Showing Improved Body Composition in Participants with Obesity
Forward split then, lol. Shelf offering! Dilution!
Yes but that is the exact opposite of a reverse split which is what you said. Reverse splits lower share count….
Increasing authorized shares can have the same end result as a split, meaning each share can be worth less, due to more shares.
Increasing authorized shares is not a reverse split ….
There is no reverse split vote!
Yes, I'm voting to reverse split.
There is no reverse split vote ….
Well, with the upcoming reverse split vote, if approved, leads to dilution, thereby pushing the stock price down.
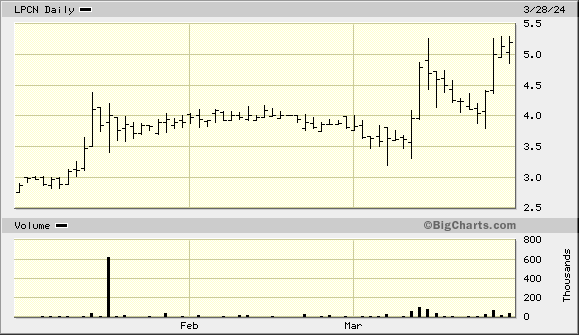
I am amazed that LPCN is now 0.92. Testosterone replacement therapy is a large market. Will sales disappoint for a reason?
"I" am always amazed when a small cap BIO gets approval for their testosterone pill over a Big Pharma that stole it and gets nailed to cross makes their stock sink?, "I" guess girls with Testosterone and Males with LPCN so no MALES in this market/world? On Watch for some males...
28 of march TLANDO pdufa, 10th march financials, this stock may grow until the approval date and over.
28 march pdufa for TLANDO and 10th sec filling am I wrong? I think this stock is going to grow constantly until 28th of march and over.
Something, nothing, anything, everything!
You said ever thing and nothing. Perfect job done.
Yes, you are correct, in March 2022, Tlando can be marketed to the public.
I remember that March 2022 is when the exclusivity for Clarus was going to end and that we could then sell our oral testosterone pills called Tlando. That was a provisional FDA decision. Is this still the case?
Thanks
If the news was good with the NASH data why is the stock flat to down?
Any time this month
Someday this stock price will either go up or go down. I'm so glad I could help.
Actually things look pretty darn good! Lawsuit aside, our pipeline is intact & progressing. TLando will still gain full approval next March, partnership news imminent (possibly soon after A/S increase),
1144 results due in a couple months with no bad expectations... yeah I'd say things are looking pretty good!
Yes, things don't look good for Lipocine or its' investors.
Correction, huge volume today.
Are you still long on LPCN?
How long have you been in LPCN?
Do you care to share your average pps or number of shares?
It appears that Lipocine has not interest in suing in court. What's the court status?
I don't think Lipocine is going to get a favorable deal for marketing Tlando. Am I being too negative? When will we hear about a partnership?
I wonder what the results of the court case are and when it will reported? Meanwhile the share price is going down, down, down, to the burning ring of fire...
It's a pretty substantial drop from 1.47 to 1.37 today. We'll see what the future holds for this.
Thanks Vance, thus far I think it's been proven that an r/s is not up for a vote, it's the dilution that's up for a vote.
Are you still long on LPCN?
How long have you been in LPCN?
Do you care to share your average pps or number of shares?
I'm not trying to be negative here, but here are some of my thoughts...
It's a shame that Tlando only got tentative approval, for marketing in March 2022, due to the fraudulent exclusivity of Jatenzo.
It appears that Lipocine has not interest in suing in court. What's the court status?
It appears that no one would want a 2 or 3 times per day Tlando pill, when Xtlando, with a once per day dose would be preferred.
It appears that no one, the FDA, patients, or big pharma has much if any interest in a T-booster pill.
I don't think Lipocine is going to get a favorable deal for marketing Tlando. Am I being too negative? When will we hear about a partnership?
Positive Nash results will be a bigger deal than Tlando, or at least a good catalyst. Unfortunately, I guess it wasn't tested on alcoholics livers, but if it treats fatty livers good, maybe it could be prescribed off label to alcoholics...
As of writing this, I'm not under water. I'm considering to bale out in full now and if I do that, I'm considering getting back in, in early 2023.
Also, the Biden Admin wants price controls/socialized medicine, which might even happen with another executive order, so lower profits for big pharma, hedge funds and us retail investors. As well as lower research capital for new drugs...
Drug costs can't just be fixed with price controls, they also need to lower NDA and SNDA costs with the FDA as well as somehow reduce the cost of drug trials.
I was under water for a,ong time on, LPCN after the FDA first reject Tlando, so I bought more and up above water now, but I mostly just see cash burning and delays here.
In the extremely long run LPCN could be a win, but if I stay in I think I'll go undwer water as the stock price tanks for the next year or so...
Am I'm being too negative and paranoid or am I being realistic. What are your thoughts?
Thank you.
You really ought to sell any shares you have in Lipocine if you believe there is any truth to your assessment.
Before or after the r/s and dilution?
|
Followers
|
50
|
Posters
|
|
|
Posts (Today)
|
0
|
Posts (Total)
|
785
|
|
Created
|
08/29/14
|
Type
|
Free
|
| Moderators | |||
675 Arapeen Drive
Suite 202
Salt Lake City, Utah 84108
Our telephone number is (801) 994-7383
Lipocine became a public company through a reverse merger in July 2013. Immediately after the reverse merger the Company raised $38 million in a private placement to institutional investors at $6.00 per share.
Lipocine pipeline products are based on its proprietary solubilization technology for effective oral delivery of water insoluble drugs to improve patient compliance.

We are a clinical-stage biopharmaceutical company focused on metabolic and endocrine disorders. We improve compliance, absorption, and more with our proprietary drug delivery technologies.
LPCN 1144, an oral prodrug of bioidentical testosterone, is being developed as a treatment for pre-cirrhotic non-alcoholic steatohepatitis ("NASH") and is currently being studied in a Phase 2 paired biopsy NASH confirmed clinical study. Liver imaging results from the Phase 2 clinical study are expected by the end of 2020 with biopsy results expected by the end of the second quarter of 2021.
TLANDO™ is an oral testosterone replacement therapy product candidate containing Testosterone Undecanoate (TU) that is designed to help restore normal testosterone levels in males for conditions associated with a deficiency or absence of endogenous testosterone.
TLANDO™ is designed to overcome many of the issues related to TRT products on the U.S. market. TLANDO is being studied for both Primary and Secondary hypogonadism and is targeting the established chronic US TRT market.
TLANDO XR is a next-generation, novel ester prodrug of testosterone which uses the patent protected Lip'ral technology to enhance solubility and improve systemic absorption. Lipocine completed a Phase 2b dose finding study in hypogonadal men in 2016. The primary objectives of the Phase 2b clinical study were to determine the starting Phase 3 dose of TLANDO XR along with the safety and tolerability of TLANDO XR and its metabolites following oral administration of single and multiple doses in hypogonadal men. The Phase 2b clinical trial was a randomized, open label, two-period, multi-dose PK study. Results suggested that the primary objectives were met, including identifying the dose expected to be tested in the planned Phase 3 study. Good dose-response relationship was observed over the tested dose range. Additionally, the target Phase 3 dose met primary and secondary end points. TLANDO XR was well tolerated with no drug-related severe or serious adverse events reported in the Phase 2b study.
Cirrhosis is an end-stage non-alcoholic fatty liver disease (NAFLD) for which there is no FDA approved drug treatment. During 2015, approximately 1.3M NASH patients had cirrhosis (fibrosis grade 4). NASH cirrhosis patients typically experience increased morbidity and symptoms of hypogonadism such as alteration of hair distribution, anemia, sexual dysfunction, testicular atrophy, muscle wasting, fatigue, osteoporosis, and gynecomastia.
Testosterone levels fall progressively with increased chronic liver disease severity. Low testosterone levels, reported in up to 90% of male cirrhotic patients, is known to increase adverse outcomes and is a predictor of mortality in these patients with increased risk of major infections, transplantation rates, increased risk of for hepatic decompensation, worsening of sarcopenia, and higher Child-Pugh score grade and MELD score. This could also include the severity of portal hypertension and ascites grade.
Our team is currently formulating plans to conduct a proof-of-concept study in male subjects with cirrhosis through consultations with the FDA and key opinion leaders to evaluate the therapeutic potential of LPCN 1148 for the treatment of cirrhosis.
LPCN 1107 is an oral product candidate of 17-alpha-hydroxyprogesterone caproate (HPC) under development for the indication of prevention of recurrent preterm birth. LPCN 1107 has the potential to become the first oral HPC product for the prevention of preterm birth in women with a prior history of at least one preterm birth. Potential benefits of our oral product candidate relative to current injectable products include the elimination of pain and site reactions associated with weekly injections, elimination of weekly doctor visits or visits from the nurse, and elimination of interference/disruption of personal, family or professional activities associated with weekly visits.
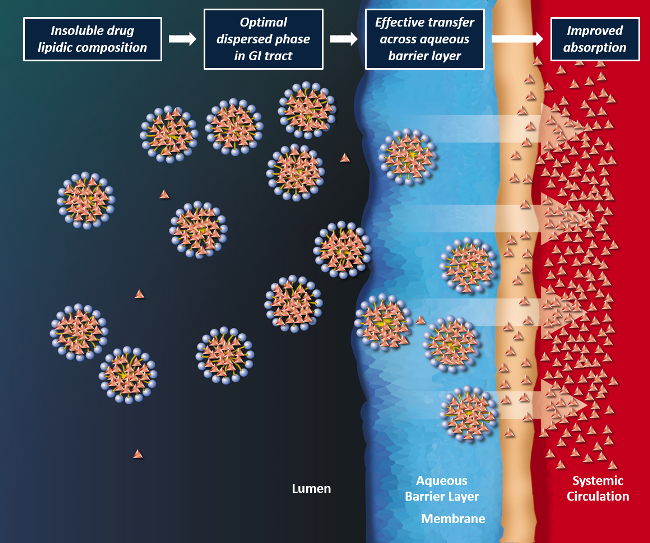
Lip'ral is a patented technology based on lipidic compositions which form an optimal dispersed phase in the gastrointestinal environment for improved absorption of the insoluble drug. Lip'ral presents insoluble drugs efficiently to the intestinal absorption site, thus bringing the absorption process under formulation control and making the product robust to physiological variables such as dilution, pH and food effects.
Non-alcoholic fatty liver disease ("NAFLD") is a reversible condition wherein large vacuoles of triglyceride fat accumulate in liver cells via the process of steatosis.
NASH is a more advanced state of NAFLD and can progress to a cirrhotic liver and eventually hepatocellular carcinoma or liver cancer. Twenty to thirty percent of the U.S. population is estimated to suffer from NAFLD and fifteen to twenty percent of this group progress to NASH, which is a substantially large population that lacks effective therapy. NAFLD/NASH is becoming more common due to its strong correlation with obesity and metabolic syndrome, including components of metabolic syndrome such as diabetes, cardiovascular disease and high blood pressure. In men, especially with comorbidities associated with NAFLD/NASH, testosterone deficiency has been associated with an increased accumulation of visceral adipose tissue and insulin resistance, which factors contributing to NAFLD/NASH.
Preclinical and clinical studies in the literature have shown the prevalence of testosterone deficiency across the NAFLD/NASH histological spectrum wherein low testosterone was independently associated with NAFLD/NASH with an inverse relationship between testosterone and NAFLD/NASH.
Post hoc analyses of existing clinical trial in subjects with comorbidities typically associated with NASH indicate that oral testosterone therapy significantly and consistently reduces elevated levels of key serum biomarkers (liver function enzymes and serum triglyceride) generally associated with NAFLD/NASH.
NAFLD prevalence in general population is estimated to be 20-30% in the Western world (Masarone et al, Rev Recent Clin Trials, 2014)
By 2020, prevalence of NAFLD cirrhosis is set to overtake hepatitis B and hepatitis C related cirrhosis (Starley et al, Hepatol, 2010)
The NASH market could peak at $30-40 billion by 2025 (Deutsche Bank industry report, “NASH – the next big global epidemic in 10 years?”, 2014)
| Volume | |
| Day Range: | |
| Bid Price | |
| Ask Price | |
| Last Trade Time: |