 News
News  Market Data
Market Data  Discover
Discover 
Support: 888-992-3836
Copyright © 2023 InvestorsHub Inc.






genisi
![]()
Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
Jaw bone created from stem cells
http://news.bbc.co.uk/2/hi/health/8290138.stm
Scientists have created part of the jaw joint in the lab using human adult stem cells.
They say it is the first time a complex, anatomically-sized bone has been accurately created in this way.
It is hoped the technique could be used not only to treat disorders of the specific joint, but more widely to correct problems with other bones too.
The Columbia University study appears in Proceedings of the National Academy of Sciences.
The bone which has been created in the lab is known as the temporomandibular joint (TMJ).
“ The availability of personalized bone grafts engineered from the patient's own stem cells would revolutionise the way we currently treat these defects ” Dr Gordana Vunjak-Novakovic Columbia University
Problems with the joint can be the result of birth defects, arthritis or injury.
Although they are widespread, treatment can be difficult.
The joint has a complex structure which makes it difficult to repair by using grafts from bones elsewhere in the body.
The latest study used human stem cells taken from bone marrow.
These were seeded into a tissue scaffold, formed into the precise shape of the human jaw bone by using digital images from a patient.
The cells were then cultured using a specially-designed bioreactor which was able to infuse the growing tissue with exactly the level of nutrients found during natural bone development.
Big potential
Lead researcher Dr Gordana Vunjak-Novakovic said: "The availability of personalised bone grafts engineered from the patient's own stem cells would revolutionise the way we currently treat these defects."
Dr Vunjak-Novakovic said the new technique could also be applied to other bones in the head and neck, including skull bones and cheek bones, which are similarly difficult to graft.
The option to engineer anatomically pieces of human bone in this way could potentially transform the ability to carry out reconstruction work, for instance following serious injury or cancer treatment.
She said: "We thought the jawbone would be the most rigorous test of our technique; if you can make this, you can make any shape."
She stressed that the joint created in the lab was bone only, and did not include other tissue, such as cartilage. However, the Columbia team is working on a new method for engineering hybrid grafts including bone and cartilage.
Another major challenge for scientists will be to find a way to engineer bone with a blood supply that can be easily connected to the blood supply of the host.
Professor Anthony Hollander, a tissue engineering expert from the University of Bristol who helped produce an artificial windpipe last year, said there was still a lot of work to be done before the new bone could be used on patients.
But he said: "One of the major problems facing scientists in this field is how to engineer a piece of bone with the right dimensions - that is critical for some of these bone defects.
"This is a lovely piece of tissue engineering which has produced bone with a high degree of accuracy in terms of shape."
On carfilzomib - I can see why Onyx is optimistic about it as its activity in heavily pretreated MM patients looks decent and adverse events are acceptable (especially since peripheral neuropathy is low grade).
ACOR/fampridine
Clearly there's going to be discussion on efficacy and the risk of seizure (which was expected). I'm more worried about the questions regarding the suitableness of the walk-test endpoint to establish the drug's efficacy and the FDA's own analysis. Still, I find the safety and efficacy profile of the 10mg BID dose approvable.
Stage Set for Acorda Bounce?
http://seekingalpha.com/article/165895-stage-set-for-acorda-bounce?source=yahoo
By M. E. Garza October 11, 2009
Shares of Acorda Therapeutics Inc. (NASDAQ:ACOR) have hit a five month low, after documents posted on the U.S. Food and Drug Administration's Website show that some agency investigators have questioned whether Acorda's proposed MS treatment Fampridine-SR should be approved.
As subscribers to the BioMedReports FDA Calendar know, a special panel is slated to meet next week to discuss whether the agency should approve the drug. The documents were posted ahead of the meeting as part of the agency's briefing materials.
So is the stage now being set for a bouce?
Some on Wall Street are still feeling bullish about the stock, despite Friday's flurry of panic driven sales.
Lazard Capital expects the FDA panel to recommend approval of Acorda's Fampridine- SR and would be a buyer of the stock ahead of meeting on October 14. The firm believes the company is well prepared for the safety questions and the firm has reiterated a "Buy" rating on the stock.
Analysts at Leerink Swann believe that the "hate selling" has happened as expected, but the firm said its confidence in Acorda is "waning" although it is maintaining an Outperform rating on the stock.
To analysts at Deutsche Bank, the FDA documents appear to be positive and the firm noted that the FDA does not appear to be worried about Amaya leading to an MS relapse. The firm maintains it's "Buy" rating on Acorda.
Acorda is seeking to have fampridine-SR approved to help improve walking speed in patients with MS. An active ingredient in the product has been suspected of triggering seizures in certain patients when used in high doses.
Interestingly, shares of Biogen Idec Inc.(NASDAQ:BIIB) traded up for the day despite the fact that it has rights to Fampridine-SR outside the U.S.
Acorda is a commercial-stage biopharmaceutical company engaged in the identification, development and commercialization of therapies that improve neurological function in people with multiple sclerosis (MS), spinal cord injury (SCI), and other disorders of the central nervous system (CNS). The Company’s marketed product, Zanaflex Capsules, is approved by the United States Food and Drug Administration (FDA) for the management of spasticity.
The Company’s product candidate Fampridine-SR, has completed two positive Phase III clinical trials for the improvement of walking ability in patients with MS.
The approval letter for Berinert says it has an orphan drug designation.
The label is weird. As far as I know, C1-Inhibitor concentrate is an effective therapy that rapidly relieves acute swelling attacks at any body location in patients with HAE. Makes sense the FDA wants 2 products in the market but I don't have a clue regarding the narrow label.
Edit: Found the reason to the weird label- Inclusion Criteriain CLS phase III trial
* Documented congenital C1-INH deficiency
* Acute facial or abdominal HAE attack
Perhaps you were thinking of Pharming's recombinant human C1 inhibitor.
Onyx to acquire private biotech Proteolix Inc for $276M
http://www.reuters.com/article/governmentFilingsNews/idUSN1212525920091012
As CSL’s Berinert is approved first for the treatment of acute HAE, it will block Cinryze from the market for this indication in the US for 7 years, due to its orphan drug status.
Re: Copaxone in CIS
The trend is to start treatment as early as possible and that means that CIS subgroup within the MS market should grow. I don't know how much of Copaxone's sales come from CIS subgroup but I assume that since both Avonex and Betaseron are approved to treat a first clinical episode with MRI findings consistent with MS, Copaxone should gain share at their expense. I haven't seen graphs but have read the info about conversions in a recent clinical review.
I don't have a better view on the CIS subgroup than what I've posted here #msg-32333213. To summarise: there's a growing body of evidence that early is late in MS — when patients develop the first symptoms, the disease has been present for a long time. Some patients may already have evidence of old lesions on MRI and some atrophy of the brain. On the other hand, it is uncertain how many would have converted from CIS to “clinically definite MS”, with a second attack in the long term, although most convert in the first five years, and especially in the first two years, after an initial attack. Physicians tend to start treatment earlier so Copaxone should gain share at the expense of the beta interferon class. Still, when dealing with those without clinical symptoms compliance can be low, discontinuations common, and some may prefer the wait-and-watch attitude.
Yes, I know he is a hopeless case but for the sake of the other two readers of the board :)
Note that Teva isn't alone there - Sanofi-Aventis, Novartis, Ranbaxy, and Ratiopharm all confirmed EU raids at French units:
http://www.reuters.com/article/rbssHealthcareNews/idUSL762567720091007
Teva/Copaxone Data in CIS
PreCISe data published in the Lancet #msg-42284652
PreCISe data published in the Lancet
http://podcast.thelancet.com/audio/lancet/2009/07october.mp3
PreCISe study and multiple sclerosis
Encouraging results from a phase III placebo-controlled trial suggest that glatiramer acetate (already licensed for relapsing-remitting multiple sclerosis) is effective in reducing progression to clinically definite disease after a first clinically isolated syndrome. Individuals were studied for three years, with MRI analysis done every 3 months to compare underlying disease progression with clinical symptoms. The study is discussed in a podcast.
http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(09)61259-9/fulltext
GPRO to acquire Prodesse Inc, a privately held provider of clinical tests for influenza and other infectious diseases for approximately $60M in cash.
http://www.reuters.com/article/marketsNews/idCNN0631723520091006?rpc=44
The ninth Israeli citizen to win a Nobel prize:
The Nobel Prize in Chemistry for 2009 jointly to
http://nobelprize.org/nobel_prizes/chemistry/laureates/2009/press.html
Venkatraman Ramakrishnan, MRC Laboratory of Molecular Biology, Cambridge,
United Kingdom
Thomas A. Steitz, Yale University, New Haven, CT, USA
Ada E. Yonath, Weizmann Institute of Science, Rehovot, Israel
"for studies of the structure and function of the ribosome"
Scientific Meeting Calendar
NOTE: ANYONE MAY UPDATE THIS FILE
Edits: Added entries for ANA, ACG, AAO, Obesity 2009
OCTOBER 2009
International Symposium on HCV and Related Viruses
October 3-7, 2009
Nice, French
http://www.hcv2009.org/
American Neurological Association - ANA
October 11-14, 2009
Baltimore, MD
http://www.aneuroa.org/
International Congress on Coronary Artery Disease - ICCAD
October 11 to 14, 2009
Prague, Czech Republic
http://www2.kenes.com/cad/pages/home.aspx
American College of Rheumatology - ACR
October 17-21, 2009
Philadelphia, USA
http://www.rheumatology.org/annual/index.asp
Society for Neuroscience - SFN
October 17-21, 2009
Chicago, USA
http://www.sfn.org/am2009/
World Diabetes Congress - IDF
October 18-22, 2009
Montreal, Canada
http://www.worlddiabetescongress.org/
American Society of Human Genetics - ASHG,
October 20-24, 2009
Honolulu, Hawaii
http://www.ashg.org/2009meeting/
American College of Gastroenterology - ACG
October 23-28, 2009
San Diego, California
http://www.gi.org/physicians/education.asp
American Academy of Ophthalmology - AAO
October 24-27, 2009
San Francisco, CA
http://www.aao.org/meetings/annual_meeting/
The Obesity Society - Obesity 2009
October 24-28, 2009
Washington, DC
http://www.obesity.org/obesity2009/
Renal Week 2009
October 27 - November 1, 2009
San Diego, CA
http://asn-online.org/education%5Fand%5Fmeetings/renal%5Fweek/
American Association for the Study of Liver Diseases (AASLD)
Oct 31 - Nov 3, 2009
Boston, USA
http://www.aasld.org/thelivermeeting/Pages/default.aspx
American College of Chest Physicians - CHEST
October 31 - November 5, 2009
San Diego, California
http://www.chestnet.org/CHEST/program/about09.php
NOVEMBER 2009
American College of Allergy, Asthma & Immunology - ACAAI
November 5-11, 2009
Miami Beach, FL
http://www.acaai.org/Member/Annual_Meeting/Annual+Meeting.htm
American Heart Association - AHA
November 14-18, 2009
Orlando, Fl
http://scientificsessions.americanheart.org/portal/scientificsessions/ss/seeyounextyear2009
DECEMBER 2009
American Epilepsy Society - AES
December 4-8, 2009
Boston, MA
www.aesnet.org
American Society of Hematology - ASH
December 5-8, 2009
New Orleans, LA
http://www.hematology.org/meetings/2009/index.cfm
International Respiratory Congresses - AARC Convention
Dec. 5–8, 2009
San Antonio, Texas
http://www.aarc.org/education/meetings/#future_congress
American College of Neuropsychopharmacology - ACNP
Dec 6-10, 2009
Hollywood, Florida
www.acnp.org
--
Procedure for Updating Calendar
When adding or modifying entries, please follow these steps:
1. Copy the complete text from the old calendar.
2. Make your additions or modifications, inserting any new items in chronological order.
3. Near the top of the message, give a very brief description of your changes (e.g. “Edits: Added entry for AASLD”).
4. Post the updated calendar in a new message as a reply to the message with the old calendar.
Glaxo kidney cancer drug wins U.S. panel support
http://www.reuters.com/article/marketsNews/idUSN0538316920091005
* FDA advisers back Votrient in 10-0 vote
* Agency will consider vote, make final decision
* Drug would be sixth one on U.S. market (Adds background on drug, comments from panel)
GAITHERSBURG, Md., Oct 5 (Reuters) -A U.S. Food and Drug Administration advisory panel gave GlaxoSmithKline's (GSK.L: Quote, Profile, Research, Stock Buzz) (GSK.N: Quote, Profile, Research, Stock Buzz) experimental kidney cancer drug Votrient its full support on Monday, saying the drug appeared effective and no more risky than other similar medications.
The drugmaker is seeking to market Votrient, also known by its chemical name pazopanib, to treat advanced renal cell carcinoma. If approved, it would be the sixth drug on the U.S. market to treat the disease.
FDA's panel of outside experts agreed with company officials that the drug would offer doctors another choice in treating patients with side effects that are somewhat different than those of its rivals.
The FDA will weigh the panel's vote before deciding whether to approve the drug.
Other approved renal cell cancer drugs include Pfizer's Sutent, Roche/Genentech's Avastin, Wyeth's Torisel, Novartis AG's Afinitor, and Onyx/Bayer AG's Nexavar.
Some of them, like Votrient, target the vascular endothelial growth factor (VEGF) receptor to try to limit new blood vessels that can feed tumors.
"They all have their pluses and minuses," said panel member Dr. Michael Kelly, an associate medical professor at Yale University. "This drug here definitely does have clinical activity, and the spectrum of side effects ... are different but may not be any more severe than the ones out there now."
Company data showed Votrient caused liver-related problems and possibly death, FDA staff told the panel.
Other side effects included diarrhea and heart problems, which Glaxo said are also seen with other VEGF drugs. Unlike rivals, Votrient led to fewer reports of skin problems and fatigue, the drugmaker said.
Panel members also urged Glaxo to complete its current study comparing its drug to Pfizer's Sutent, which has become the standard drug used in the United States. They also said the company needs to closely monitor side effects even if it wins approval.
Glaxo, in a statement, said it would continue to work with the FDA. After the vote, shares of the British drugmaker closed 0.4 percent higher at $38.86 on the New York Stock Exchange.
Whole genome sequencing technology - giant is stepping in:
I.B.M. Joins Pursuit of $1,000 Personal Genome
http://www.nytimes.com/2009/10/06/science/06dna.html?_r=3&emc=eta1
By JOHN MARKOFF, October 6, 2009
One of the oldest names in computing is joining the race to sequence the genome for $1,000. On Tuesday, I.B.M. plans to give technical details of its effort to reach and surpass that goal, ultimately bringing the cost to as low as $100, making a personal genome cheaper than a ticket to a Broadway play.
The project places I.B.M. squarely in the middle of an international race to drive down the cost of gene sequencing to help move toward an era of personalized medicine. The hope is that tailored genomic medicine would offer significant improvements in diagnosis and treatment.
I.B.M. already has a wide range of scientific and commercial efforts in fields like manufacturing supercomputers designed specifically for modeling biological processes. The company’s researchers and executives hope to use its expertise in semiconductor manufacturing, computing and material science to design an integrated sequencing machine that will offer advances both in accuracy and speed, and will lower the cost.
“More and more of biology is becoming an information science, which is very much a business for I.B.M.,” said Ajay Royyuru, senior manager for I.B.M.’s computational biology center at its Thomas J. Watson Laboratory in Yorktown Heights, N.Y.
DNA sequencing began at academic research centers in the 1970s, and the original Human Genome Project successfully sequenced the first genome in 2001 and cost roughly $1 billion.
Since then, the field has accelerated. In the last four to five years, the cost of sequencing has been falling at a rate of tenfold annually, according to George M. Church, a Harvard geneticist. In a recent presentation in Los Angeles, Dr. Church said he expected the industry to stay on that curve, or some fraction of that improvement rate, for the foreseeable future.
At least 17 startup and existing companies are in the sequencing race, pursuing a range of third-generation technologies. Sequencing the human genome now costs $5,000 to $50,000, although Dr. Church emphasized that none of the efforts so far had been completely successful and no research group had yet sequenced the entire genome of a single individual.
The I.B.M. approach is based on what the company describes as a “DNA transistor,” which it hopes will be capable of reading individual nucleotides in a single strand of DNA as it is pulled through an atomic-size hole known as a nanopore. A complete system would consist of two fluid reservoirs separated by a silicon membrane containing an array of up to a million nanopores, making it possible to sequence vast quantities of DNA at once.
The company said the goal of the research was to build a machine that would have the capacity to sequence an individual genome of up to three billion bases, or nucleotides, “in several hours.” A system with this power and speed is essential if progress is to be made toward personalized medicine, I.B.M. researchers said.
At the heart of the I.B.M. system is a novel mechanism, something like nanoscale electric tweezers. This mechanism repeatedly pauses a strand of DNA, which is naturally negatively charged, as an electric field pulls the strand through a nanopore, an opening just three nanometers in diameter. A nanometer, one one-billionth of a meter, is approximately one eighty-thousandths the width of a human hair.
The I.B.M. researchers said they had successfully used a transmission electron microscope to drill a hole through a semiconductor device that was intended to “ratchet” the DNA strand through the opening and then stop for perhaps a millisecond to determine the order of four nucleotide bases — adenine, guanine, cytosine or thymine — that make up the DNA molecule. The I.B.M. team said that the project, which began in 2007, could now reliably pull DNA strands through nanopore holes but that sensing technology to control the rate of movement and to read the specific bases had yet to be demonstrated.
Despite the optimism of the I.B.M. researchers, an independent scientist noted that various approaches to nanopore-based sequencing had been tried for years, with only limited success.
“DNA strands seem to have a mind of their own,” said Elaine R. Mardis, co-director of the genome center at Washington University in St. Louis, noting that DNA often takes a number of formations other than a straight rod as it passes through a nanopore.
Dr. Mardis also said previous efforts to create uniform silicon-based nanopore sensors had been disappointing.
One of the crucial advances needed to improve the quality of DNA analysis is to be able to read longer sequences. Current technology is generally in the range of 30 to 800 nucleotides, while the goal is to be able to read sequences of as long as one million bases, according to Dr. Church, who spoke in July at a forum sponsored by Edge.org, a nonprofit online science forum.
Other approaches to faster, cheaper sequencing include a biological nanopore approach being pursued by Oxford Nanopore Technologies, a start-up based in England, and an electron microscopy-based system being designed by Halcyon Molecular, a low-profile Silicon Valley start-up that has developed a technique for stretching single strands of DNA laid out on a thin carbon film. The company may be able to image strands as long as one million base pairs, said Dr. Church, who is an adviser to the company, and to several others.
“To bring about an era of personalized medicine, it isn’t enough to know the DNA of an average person,” said Gustavo Stolovitzky, an I.B.M. biophysicist, who is one of the researchers who conceived of the I.B.M. project. “As a community, it became clear we need to make efforts to sequence in a way that is fast and cheap.”
As an Israeli, trading from an Israeli account, for every action I do at NASDAQ, I have the currency exchange burden and the American broker' commission on top. When I buy or sell at TASE, I only pay my local broker. Until 2007 the tax on profits from NASDAQ was 25% whereas, on a stock traded at TASE only 15%.
And there's the litigation with Baxter regarding methods of screening the samples. Kamada is too small and its capacity isn't a real threat to Talecris unless of course the inhaled AAT gets approved.
It looks solid enough and Soon-Shiong may have been right all along about Abraxane.
Baxter, CSL Behring, and Talecris also compete in the A1PI market with products aimed at boosting a protein that protects the lungs from inflammation. [A1PI is another name for alpha-1 antitrypsin.] Talecris said its A1PI Prolastin product had a 67 percent share of sales in the U.S. in 2008 and 90 percent in the European Union.
Frost doesn't stop - OPKO will acquire Pharma Genexx S.A, a privately-held Chilean pharmaceutical company, for $16M in an all cash transaction, less than 4 months after it has acquired a vax development platform.
http://www.biospace.com/news_story.aspx?NewsEntityId=157894&Source=TopBreaking
I expect velaglucerase to be the most attractive option for patients not taking drug holiday during Cerezyme's shortage. I'm sure Actelion is trying to push Zavesca but I don't see many patients going on it, and I bet those who are, will switch back to Cerezyme as fast as they can. On the other hand, some of velaglucerase and prGCD switchers might stay on the alternative ERT.
Two disclosures: I'm not an expert and Biocell (the Israeli way of holding PLX) is still one of my largest positions at TASE.
Re: FOLD-Plicera Phase 2 failure
Similar to plicera, amigal is also a chaperone. I am quite skeptical about this approach as it is very difficult to design a chaperone that will restore the enzyme's function and increase the level of active enzyme.
What are your thoughts on substrate reduction therapies? Or do you think ERT is the best approach for the foreseeable future?
I never had much faith in their chaperone technology:
Amicus Therapeutics Announces Preliminary Results of Phase 2 Study with Plicera(TM) for Gaucher Disease
http://finance.yahoo.com/news/Amicus-Therapeutics-Announces-prnews-1330118971.html?x=0&.v=1
CRANBURY, N.J., Oct. 2 /PRNewswire-FirstCall/ -- Amicus Therapeutics (Nasdaq: FOLD - News) today announced preliminary results from its Phase 2 randomized, open-label study to assess the safety, tolerability and preliminary efficacy of its investigational drug, Plicera(TM) (afegostat tartrate), in treatment-naive adult patients with type 1 Gaucher disease. Two dose regimens of Plicera (225 mg three days on/four days off and seven days on/seven days off) were studied during this six month trial. While all patients enrolled experienced an increase in the level of the target enzyme (GCase) as measured in white blood cells, clinically meaningful improvements in key measures of disease were observed in just one of the eighteen patients who completed the study. The preliminary results suggest that treatment with Plicera was generally well tolerated, with no serious adverse events (SAEs) reported. Nineteen subjects were enrolled and 18 subjects completed the study. One subject discontinued treatment because of an adverse event (conjunctivitis-related symptoms).
Once the data are final, the Company plans to further analyze and evaluate the results in collaboration with its partner, Shire Human Genetic Therapies, Inc. (Shire HGT), and, based on this work, will determine the appropriate next steps for the Plicera program. However, based on these preliminary results, the Company does not expect to advance Plicera into Phase 3 development at this time.
John F. Crowley, President and CEO of Amicus, stated, "The preliminary results of this Phase 2 study certainly do not meet our expectations, but we believe they do provide additional insights into the biological activity of Plicera in Gaucher disease and the pharmacological chaperone technology platform. We plan a detailed analysis to ensure we have a complete understanding of the data."
The Company is also today announcing a change to its financial guidance for 2009. Based on its current projections of net operating expense, the Company now expects to end 2009 with approximately $70-$80 million in cash.
Conference Call and Webcast
Amicus will host a conference call to discuss the Plicera Phase 2 preliminary results, today, October 2, 2009, at 5 p.m. EDT.
snip...
Amicus Therapeutics Announces Preliminary Results of Phase 2 Study with Plicera(TM) for Gaucher Disease
http://finance.yahoo.com/news/Amicus-Therapeutics-Announces-prnews-1330118971.html?x=0&.v=1
CRANBURY, N.J., Oct. 2 /PRNewswire-FirstCall/ -- Amicus Therapeutics (Nasdaq: FOLD - News) today announced preliminary results from its Phase 2 randomized, open-label study to assess the safety, tolerability and preliminary efficacy of its investigational drug, Plicera(TM) (afegostat tartrate), in treatment-naive adult patients with type 1 Gaucher disease. Two dose regimens of Plicera (225 mg three days on/four days off and seven days on/seven days off) were studied during this six month trial. While all patients enrolled experienced an increase in the level of the target enzyme (GCase) as measured in white blood cells, clinically meaningful improvements in key measures of disease were observed in just one of the eighteen patients who completed the study. The preliminary results suggest that treatment with Plicera was generally well tolerated, with no serious adverse events (SAEs) reported. Nineteen subjects were enrolled and 18 subjects completed the study. One subject discontinued treatment because of an adverse event (conjunctivitis-related symptoms).
Once the data are final, the Company plans to further analyze and evaluate the results in collaboration with its partner, Shire Human Genetic Therapies, Inc. (Shire HGT), and, based on this work, will determine the appropriate next steps for the Plicera program. However, based on these preliminary results, the Company does not expect to advance Plicera into Phase 3 development at this time.
John F. Crowley, President and CEO of Amicus, stated, "The preliminary results of this Phase 2 study certainly do not meet our expectations, but we believe they do provide additional insights into the biological activity of Plicera in Gaucher disease and the pharmacological chaperone technology platform. We plan a detailed analysis to ensure we have a complete understanding of the data."
The Company is also today announcing a change to its financial guidance for 2009. Based on its current projections of net operating expense, the Company now expects to end 2009 with approximately $70-$80 million in cash.
Conference Call and Webcast
Amicus will host a conference call to discuss the Plicera Phase 2 preliminary results, today, October 2, 2009, at 5 p.m. EDT.
VRTX:
...if Graves had originally expected to be named the CEO when Boger stepped down
VRTX: Kurt Graves, head of commercial operations and strategic planning,
resigned today for unstated reasons.
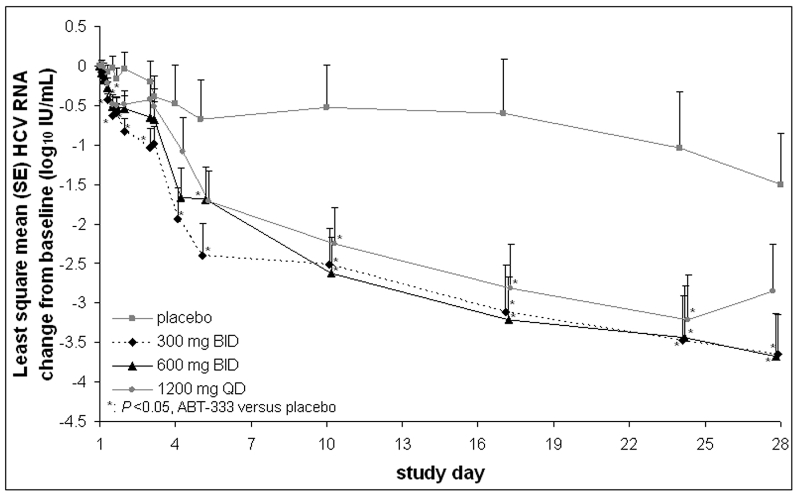
AASLD ABT-333 abstract:
Treatment-naïve, HCV genotype 1-infected subjects show significantly greater HCV RNA decreases when treated with 28 days of ABT-333 plus peginterferon and ribavirin compared to peginterferon and ribavirin alone
M. Rodriguez-Torres1; E. Lawitz2; D. Cohen3; L. M. Larsen3; R. Menon3; C. Collins3; T. Marsh3; S. Gibbs3; B. Bernstein3
1. Fundación de Investigación de Diego, San Juan, PR, USA.
2. Alamo Medical Research, San Antonio, TX, USA.
3. Abbott Laboratories, Abbott Park, IL, USA.
Objective: ABT-333 is a potent nonnucleoside HCV polymerase inhibitor with a favorable safety profile in healthy subjects. This study assesses the safety, antiviral activity, and pharmacokinetics (PK) of ABT-333 with peginterferon alfa-2a (pegIFN) and ribavirin (RBV) in HCV-infected subjects.
Methods: 30 HCV genotype 1-infected, treatment-naïve subjects were randomized to ABT-333 300 mg BID (N=8), 600 mg BID (N=8), 1200 mg QD (N=8), or placebo (n=6) for 28 days (2 days monotherapy plus 26 days with pegIFN 180 mcg/wk + RBV 1000-1200 mg/d, weight-based). Safety was monitored by adverse events (AEs) and lab results. ABT-333 PK profile was assessed on Day 1; samples were also collected Days 2, 4, 5, 10, 17, 24 and 28.
Results: Subjects were primarily male (70%) and white (90%); 43% were of Latino ethnicity. At baseline, the mean (SD) age was 46.5 (9.8) yrs and the mean weight was 78.6 (13.2) kg. Treatment with ABT-333+pegIFN/RBV resulted in statistically significantly greater decreases in HCV RNA versus placebo+pegIFN/RBV (Figure). The least square mean maximum HCV RNA change from baseline was -3.7, -4.0, and -3.5 log10 IU/mL for 300 mg BID, 600 mg BID, and 1200 mg QD ABT-333+pegIFN/RBV, respectively, compared to -1.4 log10 IU/mL for placebo+pegIFN/RBV. Mean ABT-333 Cmax and AUC increased with increasing doses and were comparable to healthy subjects. Based on mean trough values, addition of pegIFN/RBV did not alter ABT-333 PK. AEs were generally mild and attributed to pegIFN or RBV. Of ABT-333-associated AEs, nausea, headache, flatulence, and dermatitis were most common (N=2-3 for each AE). There were no serious AEs or discontinuations due to AEs. Lab abnormalities were similar in subjects receiving ABT-333 and placebo.
Conclusion: ABT-333 was well tolerated for 28 days when dosed with pegIFN/RBV and resulted in significant decreases in HCV RNA versus pegIFN/RBV alone.
ITMN-191 looks good for bid.
Safety signal for MRK's non-nuc MK-3281 - severe myoclonus. AASLD abstract:
Safety and Antiviral Activity of NS5B Polymerase Inhibitor MK-3281, in Treatment-Naïve Genotype 1A, 1B AND 3 HCV-Infected Patients
D. M. Brainard1; M. S. Anderson1; A. Petry1; K. Van Dyck1; I. De Lepeleire1; K. Sneddon1; C. E. Cummings1; R. B. Nachbar1; R. J. Barnard1; P. Sun1; P. Panorchan1; J. B. Sanderson2; E. Udezue3; F. Wagner4; M. Iwamoto1; J. Chodakewitz1; J. A. Wagner1
1. Merck & Co., Inc., Whitehouse Station, NJ, USA.
2. Chiltern (Early Phase) Limited, Dundee, United Kingdom.
3. Chiltern Place, Slough, United Kingdom.
4. Charité Research Organisation, Berlin, Germany.
Background: MK-3281 is a novel non-nucleoside hepatitis C virus (HCV) NS5B polymerase inhibitor with potent and selective in vitro activity against HCV genotypes (GT) 1a/b and 3 HCV. Safety, tolerability, pharmacokinetics (PK), resistance, and antiviral activity were assessed during multiple dose administration in HCV-infected patients.
Methods: This was a double-blind, placebo-controlled, serial panel study in 22 treatment-naïve HCV-infected patients who received 800 mg MK-3281 (17 patients: 4 x GT1b, 6 x GT3, 6 x GT1a, 1 x non-typeable [1-NT]) or placebo (5 patients) q12 hr for 7 days. All patients were followed for two weeks post therapy. Safety and resistance evaluations were performed throughout the study. Plasma samples were collected for MK-3281 PK and HCV viral RNA determination (using the Roche COBAS Taqman assay).
Results: There were no serious adverse experiences (AEs) reported. One patient discontinued due to an AE of myoclonus on study Day 2 of approximately 1.5 h in duration, preliminarily judged possibly-related to MK-3281 and rated of severe intensity. Other AEs were limited in number, transient, and rated mild to moderate in intensity with headache the most frequently reported AE. No clinically relevant laboratory safety signals were observed. However, several patients on therapy showed transient reductions in liver function tests. The PK of MK-3281 in HCV-infected patients were similar to previously reported values observed in healthy subjects. Mean maximum reductions from baseline of HCV viral RNA (SEs) were 1.3 (0.15), 3.8 (0.19), and 1.2 (0.16) log10 IU/mL for GT1a/1-NT, 1b, and 3, respectively. No on-treatment viral rebound was observed in any GT 1b patient, while 1 of 6 GT1a and 1 of 6 GT3 patients showed evidence of on-treatment viral rebound. Upon 2 week follow-up, plasma levels of HCV-RNA had returned to baseline levels in all individuals. Resistance analysis was performed on GT1a and GT1b patients.
Conclusions: MK-3281 as monotherapy for 7 days was well tolerated and demonstrated strong antiviral activity against GT1b HCV with no evidence of viral breakthrough. In vivo activity against GT1a and GT3 was limited. These findings support further clinical investigation of MK-3281 for the treatment of chronic HCV infection.
AASLD INFORM-1 abstract:
Combination therapy with nucleoside polymerase R7128 and protease R7227/ITMN-191 inhibitors in genotype 1 HCV infected patients: interim resistance analysis of INFORM-1 cohorts A-D
S. Le Pogam1; M. Chhabra1; S. Ali1; J. Yan1; M. J. Ilnicka1; H. Kang1; J. M. Wong1; A. Kosaka1; A. Ewing1; A. Seshaadri1; A. De La Rosa3; W. Z. Bradford2; K. Klumpp1; N. Shulman1; P. F. Smith1; N. Cammack1; I. Najera1
1. Roche Palo Alto LLC, Palo Alto, CA, USA.
2. Intermune, Brisbane, CA, USA.
3. Pharmasset, Princeton , NJ, USA.
Background and Aims: R7128 is a novel nucleoside polymerase inhibitor that displays a high barrier to the development of drug resistance. No R7128 resistance was observed after 2 (monotherapy) or 4 (combined with SOC) weeks. In contrast, during monotherapy with protease inhibitors such as R7227, drug-resistant variants were observed in a subset of patients, which were suppressed with SOC. Viral kinetics from INFORM1 indicated that R7128/R7227 combination effectively prevented viral rebound. The aim of this study was to monitor and evaluate the effect of this combination on the development of resistance after up to 14 days of treatment.
Methods: Baseline NS3/4A and NS5B sequence was determined for all patients in INFORM1. For cohort A, sequence encompassing NS5B and NS3/4A and/or NS3 protease (population and clonal) and phenotypic analysis of NS3 and NS5B were performed at the end of the monotherapy treatment (Day 4) and at the end of combination treatment (Day 7).
For cohorts B-D (14 day combination therapy), sequence and phenotypic studies were performed on any patient that experienced viral load rebound (≥ 0.5 log10 increase of viral load above nadir).
Results: 48 of 49 patients receiving the R7128/R7227 combination had a continuous viral load decline on treatment.
In cohort A, population and clonal sequence and phenotypic analysis showed no evidence of resistance.
In cohorts B-D, 1 patient had a 1.4 log10 IU/mL increase in viral load from nadir. Sequence and phenotypic analysis of the NS3 region showed no evidence of R7227 resistance. Viral load for this patient remains undetectable after 12 weeks of SOC.
Baseline population sequence of one patient receiving R7128/R7227 revealed the presence of E168 in NS3; an amino acid associated with R7227 resistance. This patient experienced a continuous viral load decline on R7128/R7227 treatment (viral load of 139 IU/ml at day 14 ). Complete analysis of NS3/4A, NS3 protease and NS5B regions will be reported.
Conclusions:
Low dose combination therapy of the nucleoside polymerase inhibitor R7128 (that presents high barrier to resistance) with the protease inhibitor R7227 (that presents potent anti-viral activity and a lower barrier to resistance) achieves rapid and sustained antiviral activity without apparent selection of resistance for up to two weeks of treatment. The ability of this DAA combination to reduce HCV viral load even in the presence of pre-existing R7227 resistant variants indicates that this particular drug combination may have unique attributes relative to other combination strategies where two drugs with lower resistance barrier are combined.
Boceprevir in null responders:
Response-Guided Therapy (RGT) for Boceprevir (Boc) Combination Treatment? – Results from HCV SPRINT-1
Background: HCV SPRINT-1 investigated a 4-week lead-in of PegIntron (P;1.5 mcg/kg/QW) plus Ribavirin (R;800-1400 mg/day) prior to the addition of Boc (800 mg TID) for 24 or 44 weeks. Analysis of this data may lead to RGT paradigms.
Methods: Viral response was assessed by Roche TaqMan (LLD=15 IU/ml) at multiple time points including treatment weeks 4, 8, 12, 24 and 24 weeks post-treatment (sustained virologic response; SVR). Results: Patients were all G1 (1a>1b) with 15% African-Americans, 7% cirrhotics and 90% high viral load. W8 virology was available for all 103 patients in each arm. The majority of patients (64%) became negative by week 8 and SVR rates were similar for the long (94%) and short (82%) treatment arms (p=NS). In contrast, patients who first became negative between week 8 and 16, benefited from longer therapy (SVR 79% vs 21%; p=0.004), but represented only 18% of the population. A third group never achieved undetectable HCV-RNA by W16; this group primarily comprises null responders (11/18 in 48W arm) at week 4.
Conclusions: The majority of patients (64%) had undetectable HCV-RNA after 4 weeks of triple therapy following the lead-in and had a high rate of SVR (82%) following a shortened 28-week treatment duration. Only 18% of patients first achieving undetectable HCV-RNA after week 8 and before week 16 of therapy benefited from a longer treatment regimen of 48 weeks. These data suggest that only a minority of treatment-naïve G1 patients will require more than 28 weeks of therapy, and response-guided therapy based on week-8 viral response may be a powerful predictive tool to individualize therapy. The SPRINT-2 trial is designed to prospectively confirm this treatment paradigm.
ghmm, on the last action, I meant after you choose "Browse", you get the dates and look for abs. Got it?
Boehringer Ingelheim's BI 201335 poster presented at AASLD 2009
Virological Response and Safety of BI 201335 protease inhibitor, Peginterferon alfa 2a and Ribavirin treatment of HCV genotype-1 patients with compensated liver cirrhosis and non-response to previous peginterferon / ribavirin
Background: BI 201335 is a highly potent and specific HCV NS3/4A protease inhibitor. A phase 1 trial in treatment-experienced HCV GT-1 patients demonstrated a mean viral load (VL) reduction of 5.3 LOG10 (IU/mL) for BI 201335 given once daily after 28 days in combination with peginterferon alfa (PegIFN) 2a and ribavirin (RBV). We now describe a phase 1b trial which has assessed safety, short-term efficacy, and pharmacokinetics of BI 201335 in GT-1 patients with compensated liver cirrhosis and non-response to previous PegIFN/RBV, a difficult-to-treat HCV population with a high unmet medical need.
Methods: In this open-label, sequential group comparison, HCV GT-1 patients with compensated liver cirrhosis who have never achieved undetectable VL under previous PegIFN/RBV were treated with 240 mg once (QD; n=6) or twice daily (BID; n=7) in combination with PegIFNa2a (180 mcg/week) and RBV (1000/1200mg/d) for 28 days. All patients received a single loading dose of 480mg of BI 201335 as the first dose. Plasma HCV RNA was measured by Roche COBAS TaqMan assay.
Results: Mean age was 54 years, BMI 26 kg/m2. Mean VL at baseline was 6.1 and 6.3 LOG10 (IU/mL) in both groups. All patients showed a rapid and continuous decline in VL. Mean VL declines on day 28 in the 240mg QD and BID groups were -4.9 and -5.0 LOG10, respectively. No breakthrough (>0.8 log rebound from VL nadir) was observed during treatment. At day 28, 5/6 and 5/7 patients achieved VL below level of quantification (< 25 IU/ml) in the QD and BID group. Furthermore, 4/6 and 1/7 patients had VL below level of detection (<10 IU/ml) in the 240mg QD and BID groups. There were no SAE in the 240mg QD group and 2 SAE in the 240mg BID group. Both were cases of mild to moderate hepatic decompensation attributed to PegIFN/RBV by the investigators. Two patients in the BID group discontinued treatment early, one due to nausea, one due to hepatic decompensation (SAE). Jaundice due to isolated unconjugated hyperbilirubinemia was reported in 2/6 and 1/7 patients at 240mg QD and BID, respectively. Other AE were mainly mild to moderate and typical of PegIFN/RBV. Lab analyses showed decreases of ALT / AST as well as blood cell counts typical of PegIFN/RBV.
Conclusions: BI 201335 once or twice daily combined with PegIFN/RBV exhibited potent antiviral activity in non-responder patients with liver cirrhosis. BI 201335 also exhibited a good safety and tolerability profile in these patients, allowing for their inclusion into the ongoing phase 2 program. These data also confirm that IFN non-responsiveness in previous non-responders can be overcome by rapid and profound inhibition of viral replication by BI 201335.
Go to www.aasld.org then to The liver meeting tab, then to abstracts and presenters, then to itinerary planner, then to browse by dates.

