News
News  Market Data
Market Data  Discover
Discover 
Support: 888-992-3836
Copyright © 2023 InvestorsHub Inc.


Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
They are probably a shorter. The lowest form of humanity. These people bankrupt companies and kill jobs for others who work in these companies that get their stocks shorted. They thrive on wickedness and evil. And get joy and jollies from hurting others.
If your book only contained all your post on I hub it would be about 500 pages
Not exactly certain, but I am holding for the results. I almost sold some shares for some pocket money but held off. Don't want to miss any news. My luck is I sell and then news hits and it takes off. My CA is $1.29 per share. No skin off my back with the drop.
At some point, these biotechs need to prove they have promising drug candidates in clinical trials and are not just good at raising capital
When is next fda news jan19?
yahoo finance says $41 price in 1 year $$$$$$$$$$$
This thing could be like Bill Bixby. You know the 1978 series whereby just before he gets extreme stress and anger. Holding for next year+++.
Check it out now. Glad I held. Very low float. Nice gem I was tipped off about and looking forward to the FDA approvals.
MONSTER BIG BUYS 15000 AND 14000 SHARES. $30K EACH BUY
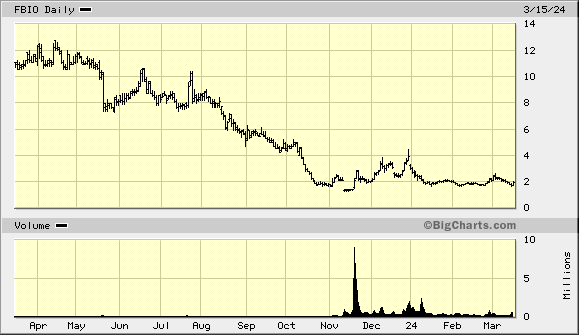
Shorts kept it in check. Profit takers made it drop. No volume for them to brag about. So the drop is normal business as usual. If you see a large drop AND very large abnormal volume... that is when to worry. Remember insiders bought this stock at an average of $9.00 + per share pre R/S. They know it is going into the $40's next year... Nice 20 bagger.
FBIO PUSHING UP IN PRE, LETS SEE THAT 3 DOLLA
Stick to your hobby of posting 52 week high and 52 week lows chief.
FBIO HOLY FUC!!!
FBIO FAB!!!
Chief keep posting your 52 week highs and lows. Get a better hobby
Then don't waste your time here. No one needs anyone to protect them from themselves. We have enough with gubberment. Speaking of which should be abolished.
It’s a wash trade event
You’re pushing the envelope
That is fine, but this one is looking promising as time goes on. Got to be in it to win it. FDA rubber stamps approvals these days. Years ago it was a different story. They relaxed most standards, so not worried at all. late 2020 and 2021 proved it with the CLOVER schots. FDA is nothing but a paid co-conspirator to bad. So use it to your benefit.
FBIO...NEXT STOP 2.27
FBIO...MONSTER
Biomed stocks and med device stocks are tricky
The sectors have a combined count of close to a thousand
Not all, in fact no more than 5% will be long winners
Yes indeed. Better than CVM where I have been stuck since 2008 or so. Still down over 80% of my initial investment of $900 or thereabouts.
FBIO...HERE WE GO
Yep and I bought up shares. Way lower than company owners bought at for sure.
Are you taunting? lol ...
Hi my friend..adding FBIO$ today yr low..tough crowd, gl...
Has FBIO gotten a NASDAQ warning letter yet?
I like the company, but needs to get over $1.00
|
Followers
|
28
|
Posters
|
|
|
Posts (Today)
|
0
|
Posts (Total)
|
211
|
|
Created
|
07/01/15
|
Type
|
Free
|
| Moderators | |||
| Volume | |
| Day Range: | |
| Bid Price | |
| Ask Price | |
| Last Trade Time: |