 News
News  Market Data
Market Data  Discover
Discover 
Support: 888-992-3836
Copyright © 2023 InvestorsHub Inc.





Register for free to join our community of investors and share your ideas. You will also get access to streaming quotes, interactive charts, trades, portfolio, live options flow and more tools.
We’ll call it a draw! You were right about this piece of garbage and I was right about the ACB piece of garbage. Truce and peace wherever you are…
Been over 18 months since i checked in last. Same as it ever was. CRICKETS!
Back up in the tree i go.
ABM
Does it mean we all ILNS investors lost all our money and we all will not have a chance to Recoup our money back ?? Or other chance to screw and put Maza and his gangsters in jails ??
No, it’s Aurora. And they’ve now written off 1.5 BILLION of the Goodwill in just the last 6 months ending 12/31/19 - WELL BEFORE all this market turmoil. The details are below -
https://investorshub.advfn.com/boards/read_msg.aspx?message_id=154303443
But here’s the direct link from the Company. The gory details are all in there -
https://investor.auroramj.com/investor-info/financial-reports/
.Chain and Maza should be in jail right now?
What have you all talked about? Here is Ilns message ?
Anyone have and know what is going with ilns ? What we can do to recoup our money back? Chain and Maza should be in jail right now?
Goodwill is their biggest asset? What planet are you looking at? Convertible debt?
Assets
Current
Cash and cash equivalents 152,526
Intangible assets 682,694
Goodwill 3,173,006
Total assets 5,606,799
Current
Accounts payable and accrued liabilities 163,579
Convertible debentures 248,617
Loans and borrowings 25,977
Contingent consideration payable 22 25,411
Convertible debentures 264,806
Convertible debentures 264,806
Loans and borrowings 256,328
Total liabilities 1,140,389
management concluded that the carrying value of the cannabis operating segment was higher than the $3.9 billion recoverable amount
and recorded impairment losses of $762.2 million during the three and six months ended December 31, 2019 (three and six months ended
December 31, 2018 - nil). The impairment was allocated entirely to reduce goodwill for the cannabis operating segment. The impairment loss was
recognized due to a change in overall industry/market conditions, a change in management’s forecasted sales and profitability outlook and a
realignment and refocus of strategic plans to meet market demand.
Wtf I believe there are 5 or 600 institutional holders.
They have an interim CEO, so in your mind they won't hire a high flying CEO even though they are actively looking to hire one?
Lol, sit down., sheesh
I misspoke. They are not the only producer, but the only Canadian owned producer/ supplier. There is also Canopy. They have entered their asset as " goodwill" so you may be confusing the two. They've tapped the convertible market as their finance facility of choice. They have some fairly large balanc sheet problems, and I suspect they'll likely be acquired, perhaps even by Aurora down the road. I don't know that much about them however, and it was over a year ago I looked into them and decided that ACB was a much better bet.
They now have 2 unimpressive quarters out which basically shocked the street
The good news is that the balance sheet is relatively strong and other players in the sector are dropping like flies.
Long term, say 5 years out, I think they'd need to really screw up not to have a +$100 PPS If they bring in a high profile CEO, it would triple from the current $1.00 PPS virtually overnight.
Lots to consider.. the first run up was a blast, and likely not the last.
Dude it's the stock market. The company dropped the ball in a bad way. They now have 2 unimpressive quarters out which basically shocked the street
I think in another year or so once retail stores are abundant, they'll be rewarded. Until then it's going to be a shit show and may test 50 cents. The good news is that the balance sheet is relatively strong and other players in the sector are dropping like flies. Long term, say 5 years out, I think they'd need to really screw up not to have a +$100 PPS If they bring in a high profile CEO, it would triple from the current $1.00 PPS virtually overnight.
Lots to consider.. the first run up was a blast, and likely not the last.
After the verdict ( settlements) , what we all ilns investors can do / go from here to get/ recoup our Hard earned money back from these crooked greedy people?
Did you get any money yet from the settlements ?
Please inform Nancy Brown, the lead lawyer for the SEC in the Team Honig case.
BrownN@sec.gov
Can we ILNS investors and our lawyers Call SEC to complain about what Maza , Chain ... have scammed and stolen very hard earned money from innocent people and seize their assets to return money to innocent people??
This con man Elliott should be in jail forever now??How much hard money has he stolen from innocent people around the world, specially in USA ?
David Blech is co founder in Tau Bio-Logic Corporation, now tauc3 biologic limited.
He and Dr. Chain and Maza shut Down Ilns to take the most promissing science and patents private - TAUC3 ANTIBODY
I was wondering why that photo looked so damned familiar...it's him, without the makeup - or not as much, anyway.


Hopefully you took my advice; ACB is now over ten bucks, a double from when I told you. Read the rest because it's not even close to being too late to get in. At the rate it's moving, 100 bucks is plausible
At this point, buddy, I don’t even know that God would jump in to make that true, although there are 10 days to go. Never give up hope - in anything.
Good luck and take care. Godspeed.
Hopefully you took my advice; ACB is now over ten bucks, a double from when I told you. Read the rest because it's not even close to being too late to get in. At the rate it's moving, 100 bucks is plausible this year.
Bought some yesterday! I figured at $2.58 now that if it goes to $100.00 by 12/31, we’ll both be IN DA MONEY! Fingers crossed!
Hopefully you took my advice; ACB is now over ten bucks, a double from when I told you. Read the rest because it's not even close to being too late to get in. At the rate it's moving, 100 bucks is plausible this year.
Look at ACB at 5 or 6 bucks. It will double this year, and in a few years liking North of 100 bucks.
Look at ACB at 5 or 6 bucks. It will double this year, and in a few years liking North of 100 bucks.
Hopefully you took my advice; ACB is now over ten bucks, a double from when I told you. Read the rest because it's not even close to being too late to get in. At the rate it's moving, 100 bucks is plausible this year.
what will we do to recoup our money back? go to court?
Dr. Screwed ILNS Shareholder with TAUC3
https://taubiologic.com/management/
Hopefully you took my advice; ACB is now over ten bucks, a double from when I told you. Read the rest because it's not even close to being too late to get in. At the rate it's moving, 100 bucks is plausible this year.
Good luck.
So the moral of the story is?
I'd say that instead of pumping this POS laden with crooks and scams, as I've pointed out for over a decade, you should have been taking notes and and performing actual due diligence, instead of spouting all the nonsense that this scam company was trying to lay on people. So here you are doing due diligence when it's far too late after you helped get other suckers into this disaster.
Here's some more advice, the last you'll get from me.
Move on, get over it, stop internalizing it, and enjoy your life. You might also want to consider not investing in this manner, because you are really bad at it; perhaps it's due to your difficulty with English, I don't know.
Might be a good idea to look at some big IPO's coming up, Uber, or take a look at the cannabis sector, something I mentioned way back here in 2008. Look at ACB at 5 or 6 bucks. It will double this year, and in a few years liking North of 100 bucks.
Enough time wasted here already, move on..
Alpha Capital fraudulent Martin Shlomo Schlaff and fraudulent Jesselsons, Honig, Maza
Stocks manipulation, stealing assets...
ILNS (HONIG,SCHLAFF,MAZA,JESSELSON,...)
MGTI
PTE
RIOT
BIOZONE
...............
https://www.haaretz.com/israel-news/.premium-interior-minister-dery-hasn-t-learned-from-the-past-1.6675204
https://en.globes.co.il/en/article-370042
https://www.haaretz.com/1.4711623
https://www.addendum.org/kern/schlaff-alpha-capital/
https://www.marketwatch.com/story/sec-charges-multiple-firms-individuals-with-microcap-fraud-schemes-including-barry-honig-2018-09-07
https://www.miaminewtimes.com/news/miami-billionaire-phillip-frost-sued-for-additonal-stock-fraud-10971262
Been writing it off for two years now two years to go.
i think now you can use the ilns loss against any gain you have in the future
You beat me to it as usual Renee!
Well that's it for ILNS, they are revoked. Mr.Maza thanks you for donating so much to him, as does David Bletch and all the rest of the scammers that brought this company to you!
LeGiOn batting 1000, bye bye =)
ILNS: registration revoked:
https://www.sec.gov/litigation/admin/2018/34-84693.pdf
you cant sell your stock no one buying ,best bet is to hope they file bankruptcy and they go belly up when that shows you can write off the loss against any gain i have a pot stock clsh made a good gain and believe im gonna make a lot larger gain in the next couple yrs so when i sell clsh ill write off the gain against the ilns losswont owe any taxes
|
Followers
|
168
|
Posters
|
|
|
Posts (Today)
|
0
|
Posts (Total)
|
20775
|
|
Created
|
10/14/08
|
Type
|
Free
|
| Moderators | |||
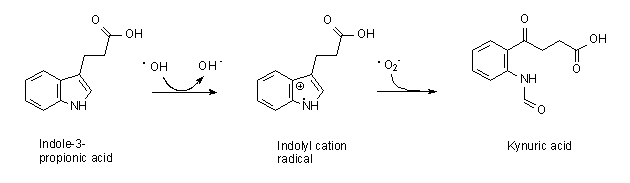
In 2011, we granted an exclusive license to ViroPharma Inc. (now part of Shire plc) covering use of our drug candidate, “OX1” (renamed by Shire as SHP622), and certain of our licensed patents and patent applications related to OX1. Shire is developing SHP622 for the treatment of Friedreich’s Ataxia (“FA”). This drug candidate is a naturally occurring small molecular weight compound (indole-3-propionic acid) that prevents oxidative stress by a combination of hydroxyl radical scavenging activity and metal chelation.
Phase 1 studies in healthy adults were completed in 2010. The drug was found to be generally well tolerated, and the pharmacokinetics revealed that the drug was rapidly absorbed and distributed in the body after oral administration.
Recently, Shire completed a Phase 1b trial to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of SHP622 in 55 adults with FA. SHP622 was generally safe and well tolerated when administered as single and multiple ascending doses. There were no severe treatment emergent adverse events (“TEAEs”) or deaths reported in either the single or multiple dose groups, and the majority of TEAEs were of mild severity. Overall, there were no clinically meaningful differences between SHP622 and placebo or between the single and multiple dose groups. The mean terminal elimination half-life ranged between 7.36 and 10.33 hours across all dose groups. Inter-subject variability appeared to be low to moderate.
Shire has stated that it will continue to analyze the results from the recently completed study and determine an optimal path forward for this program.
| PHASE 1B SAFETY, TOLERABILITY, PK/PD STUDY OF SHP622 IN ADULTS WITH FA | |
|---|---|
| TRIAL DESIGN | Randomized, Double-blind, Placebo-controlled, Multicenter, Single and Multiple Ascending Dose Studies |
| ARM 1 | Experimental: Single dose of SHP622 or placebo
|
| ARM 2 | Experimental: Multiple doses of SHP622 or placebo
|
| ENROLLMENT | 55 |
| COMPLETION | Last patient was enrolled in June 2015; Formal study report completed in April 2016 |
| PRIMARY OBJECTIVE | Evaluate the safety and tolerability of single and multiple oral doses of SHP622 in subjects with FA |
| SECONDARY OBJECTIVE | Characterize the pharmacokinetics of SHP622 by investigation of the plasma concentration-time profile following single and multiple oral doses |
| EXPLORATORY OBJECTIVE | Investigate the pharmacodynamic effects of SHP622 on plasma 8-isoprostane and malondialdehyde and urinary 8-hydroxydeoxyguanosine concentrations following multiple oral doses |
| RESULTS | SHP622 was generally safe and well tolerated when administered as single and multiple PO doses. There were no severe treatment emergent adverse events (“TEAEs”) or deaths reported in either the single or multiple dose groups, and the majority of TEAEs were of mild severity. |
| NEXT STEPS | Shire is determining the optimal path forward for this drug candidate. |


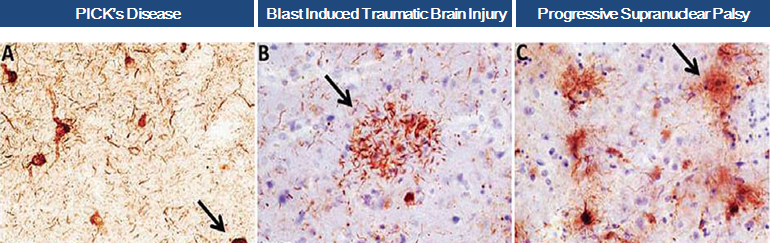
In 2012, we obtained from Northwestern University an exclusive license to worldwide diagnostic and therapeutic applications of a novel monoclonal antibody targeted to caspase cleaved TauC3.
Scientific research and pre-clinical experiments have demonstrated that fragments of Tau protein are present in brains of patients suffering from various neurodegenerative diseases, commonly known as “tauopathies”, and that the truncated form of Tau, known as TauC3, is especially toxic.
We have conducted experiments to demonstrate that caspase cleaved TauC3 is present in models of various tauopathies at a level detectable by our anti-TauC3 antibody and that our anti-TauC3 antibody shows sufficient binding specificity to the target TauC3 protein to indicate that the antibody may be a viable treatment agent.
In 2014, we reported positive top line data from a pre-clinical study conducted on our behalf at UC Irvine showing proof of concept for the antibody in an Alzheimer's disease (AD) mouse model.
In April 2016, we obtained positive data from an in-vitro study conducted on our behalf at Harvard Medical School and the MassGeneral Institute for Neurodegenerative Disease. The experiments demonstrated that our anti-TauC3 antibody significantly blocks TauC3 fragment seeding, aggregation and toxicity, demonstrating that caspase cleaved TauC3 is a potential target for treatment in Progressive Supranuclear Palsy, Traumatic Brain Injury and other tauopathies and that our antibody may be a viable treatment agent.
We are initiating experimental treatment trials of our anti-TauC3 antibody in mouse models of progressive supranuclear palsy (PSP) and traumatic brain injury (TBI). These conditions are rare, “orphan”, neurological diseases of high unmet medical need with no approved therapies.
Scientific evidence has proven that caspase cleaved TauC3 promotes formation of neurofibrillary tangles (NFTs), which are aggregates of hyper-phosphorylated tau found in numerous tauopathies.
Truncation of tau by caspases may occur relatively early in neurodegenerative disease, prior to the key folding steps that lead to the formation of phosphorylated pathological forms of tau. Also, it has been demonstrated that tau truncated at Asp421 (TauC3) aggregates more rapidly and to a greater degree than full-length tau.
Collectively, these studies suggest that the caspase cleavage of tau propagates the formation of NFTs and facilitates filament formation, which are disease- triggering events that are associated with various neurodegenerative conditions, such as Alzheimer’s disease, PSP and TBI.
We are conducting research into the use of antibody drug conjugates (ADCs), as a treatment of amyloidosis and other types of proteinopathies. ADCs chemically combine two different molecules - an antibody and a small molecule - into a single entity. We refer to this approach as “CONJUMAB”.
Our initial ADC candidate, CONJUMAB-A, is designed to combine an amyloid beta monoclonal antibody with a derivative of melatonin, thereby combining the amyloid clearing properties of the antibody with a potent neuroprotectant molecule. The rationale is that the antibody clears the amyloid peptide while the small molecule reduces the neurotoxicity caused by the peptide and provides additional neuroprotective effects to the retina.
CONJUMAB-A is an ideal compound to target amyloid beta in the eye, believed to be the leading cause of Age-Related Macular Degeneration (AMD). CONJUMAB-A would remove this toxic molecule and defend against the presence of free radicals.
We purchased the amyloid beta monoclonal antibody from Immuno-Biological Laboratories Ltd and collaborated with London-based Medical Research Council Technology to create a humanized form, which has the same properties as the original antibody.
CONJUMAB patents are pending worldwide.
The following description of Friedreich’s ataxia (FA) may be found on the website of FARA, the Friedreich’s Ataxia Research Alliance, at wwww.curefa.org.
Friedreich’s ataxia (FA) is a debilitating, life-shortening, degenerative neuro-muscular disorder. Patients suffer progressive degeneration of the central and peripheral nervous systems, which causes impaired motion and gait; diminished vision, hearing and speech; loss of strength and coordination, leading to wheelchair use; increased risk of diabetes; and life-threatening heart complications. About one in 50,000 people in the United States have Friedreich's ataxia.
Most individuals have onset of symptoms of FA between the ages of 5 and 18 years. Adult or late onset FA is less common, <25% of diagnosed individuals, and can occur anytime during adulthood.
FA is an inherited or single gene disorder. Mutations or DNA changes in the FXN gene cause FA.
FA in inherited in an autosomal recessive manner, meaning that individuals with FA have two mutated or abnormal copies of the FXN gene, this means both biological parents must be a carrier of the disease for a child to be affected. It is estimated that 1 in 100 people are carriers, and carriers do not exhibit symptoms of FA. Each such carrier parent has one mutated gene (allele) and one normal gene (allele) in the FXN gene. Because each child gets one of the mother’s genes and one of the father’s genes in this location, there are four possible combinations of the genes passed down to the child or a 25% chance that the child will have FA.
The FA gene mutation limits the production of a protein called frataxin, which is known to be an important protein that functions in the mitochondria (the energy producing factories) of the cell. Frataxin helps to move iron and is involved with the formation of iron-sulfur clusters, which are necessary components in the function of the mitochondria and thus energy production. We also know that specific nerve cells (neurons) degenerate in people with FA, and this is directly manifested in the symptoms of the disease.
Shire is developing SHP622 as a treatment for FA on the basis that SHP622’s radical scavenging activity and metal chelation properties counteract the excess amount of iron in the mitochondria and free radicals in the bodies of FA patients.
There is no FDA approved treatment for FA; at typical orphan drug prices, FA represents a target market with worldwide peak year sales of ~ $1B ($440M-$770M in US and EU alone).
Penetration of the FA market should be rapid because of the significant unmet medical need and lack of competition.
Progressive Supranuclear Palsy (PSP) is a progressive brain disorder that resembles Parkinson’s disease. PSP affects movement, control of walking (gait) and balance, speech, swallowing, vision, mood and behavior, and thinking. Classic signs of the disease are an inability to aim and move the eyes properly and blurring of vision. Symptoms begin on average after age 60 and men are affected more often than women. It is estimated that PSP affects 6 in 100,000 Americans; Incidence 2.8 per 100,000.
The exact cause of PSP is unknown. The symptoms of PSP are caused by a gradual deterioration of brain cells in specific areas in the brain, mainly in the region called the brain stem. The hallmark of the disease, and a possible cause, is the accumulation of abnormal deposits of tau and eventual toxicity in nerve cells in the brain stem. These findings suggest that the use of tau antibody therapeutics, such as such as our anti-TauC3 monoclonal antibody, may be a viable treatment approach.
There is no FDA approved treatment for PSP. No medication is effective in halting the progression of the disease; however, several medications, including dopamine agonists, tricyclic antidepressants, and methysergide, may provide modest symptomatic improvement.
Drugs in development for PSP include: BMS-968168 (Bristol-Myers Squibb) and ABBV-8E12 (AbbVie) both of which are anti-tau antibodies in Phase 1; and several drugs in preclinical stage development.
Traumatic Brain Injury (TBI) is a form of acquired brain injury, which occurs when a sudden trauma causes damage to the brain. Sports-related concussion, which includes chronic traumatic encephalopathy (CTE) is caused by direct impact forces to the head; blast-induced TBI is caused by exposure to blast shock waves.
TBI results from secondary neuronal damage caused by the impact, which leads to progressive neuronal cell death, neural loss, and axonal degeneration in the brain. Symptoms of TBI can be mild, moderate, or severe, depending on the extent of the brain damage. Patients suffer headache, light-headedness, memory loss, confusion, attention deficits, difficulty balancing, aggression, anxiety, depression, etc.
The CDC estimates that TBI affects approximately 1.7 million Americans each year, including approximately 270,000 NFL players and 200,000 war veterans.
There is no FDA approved treatment for TBI; moderately to severely injured patients receive rehabilitation that involves treatment programs in physical therapy, occupational therapy, speech/language therapy, physical medicine, psychology/psychiatry, and social support.
A common feature of TBI related diseases is deposition of the tau protein around cerebral blood vessels in the frontal cortex of the brain. Studies have indicated that military personnel who reported three or more traumatic brain injuries showed high total tau protein concentrations in plasma, in some cases long after the injuries had occurred. These findings are consistent with reports for repetitive head injury in athletes linked to progressive tauopathy, axonal injury and post-concussive disorder symptoms. These findings suggest that the use of tau antibody therapeutics, such as our anti-TauC3 monoclonal antibody, may be a viable treatment approach.
Age-Related Macular Degeneration (AMD) is the leading cause of blindness in elderly people. The global market for AMD is estimated at $4 billion annually.
There are two types of AMD: Wet AMD, characterized by new blood vessel formation; and Dry AMD, with precursor characterized by drusen and atrophic loss of retinal pigment epithelium.
Dry AMD is a large potential market. Approximately 80 percent of patients with AMD have the dry form. Central loss of the macula and underlying retinal pigment epithelium, termed GA, is considered the most severe form of Dry AMD and is responsible for over 20 percent of all cases of legal blindness in North America. There is no approved treatment existing today for Dry AMD.
Evidence from preclinical studies on retinal degeneration demonstrate that combining treatments targeting different components of the amyloid beta formation and aggregation pathway is more effective than monotherapy, indicating that CONJUMAB-A may be an effective treatment agent for Dry AMD.
| Market Value1 | $122,475 | a/o Jul 05, 2016 | |
| Authorized Shares | 2,000,000,000 | a/o Nov 04, 2015 | |
| Outstanding Shares | 5,567,037 | a/o Nov 04, 2015 | |
| -Restricted | Not Available | ||
| -Unrestricted | Not Available | ||
| Held at DTC | Not Available | ||
| Float | 4,717,663 | a/o Nov 04, 2015 | |
| Par Value | Not Available |
Capital Change=shs decreased by 1 for 50 split Pay date=04/12/2011. |
| Volume | |
| Day Range: | |
| Bid Price | |
| Ask Price | |
| Last Trade Time: |





